Decoding Genetic Circuit Failures: Analyzing Static and Dynamic Failures in Genetic Circuitry
Synthetic biology resides at the nexus of engineering and biology, employing diverse ap- proaches to engineer biological systems. These systems can be as simple as DNA sequences, bio- chemical reactions, or more abstracted through control theory or digital logic, among other ways. Similar to other engineering disciplines, for real-world applications, the designed systems must ex- hibit robustness and adaptability to environmental changes beyond controlled laboratory settings. This dissertation focuses on genetic constructs viewed specifically as digital logic genetic circuits, examining their implementation and failure behavior. It aims to elucidate and analyze various failure modes and proposes analytical methods to enhance genetic circuit robustness. This work defines genetic circuit failure, where deviations from expected output are deemed as unexpected and faulty. Such deviations may stem from failures at the cellular level or from flaws in the circuit’s logic implementation or Boolean function. Subsequently, this dissertation develops computational methods to predict circuit behavior, employing diverse analysis techniques such as ordinary differ- ential equation analysis, stochastic simulation algorithms, and stochastic model verification. These methodologies enable the prediction of the likelihood of failure occurrence. Furthermore, this dis- sertation compares different computational modeling techniques to assess the effort required for genetic circuit analysis. Finally, experimental validation is provided for a predicted circuit failure, demonstrating the practical application of the proposed methodologies.
SeqImprove: Machine-Learning-Assisted Curation of Genetic Circuit Sequence Information
The progress and utility of synthetic biology is currently hindered by the lengthy process of studying literature and replicating poorly documented work. Reconstruction of crucial design information through post hoc curation is highly noisy and error-prone. To combat this, author participation during the curation process is crucial. To encourage author participation without overburdening them, an ML-assisted curation tool called SeqImprove has been developed. Using named entity recognition, called entity normalization, and sequence matching, SeqImprove creates machine-accessible sequence data and metadata annotations, which authors can then review and edit before submitting a final sequence file. SeqImprove makes it easier for authors to submit sequence data that is FAIR (findable, accessible, interoperable, and reusable).
Rare-Event Guided Analysis of Infinite-State Chemical Reaction Networks
Probabilistic Model Checking (PMC) is a valuable tool for automated analysis of systems exhibiting stochastic behavior. However, the effectiveness of PMC algorithms is limited to systems that can be modeled by a finite state-space. Chemical Reaction Networks (CRNs) are commonly used to describe biochemical systems. Since there are usually no upper-bounds on the population of species in a CRN, they can only be modeled as an infinite-state stochastic model. This paper proposes a new approach that can analyze infinite-state CRNs by bounding their state-space. For a property indicating that the probability of the event of interest is less than a certain threshold value, the objective is to generate a bounded range on the population of each species in the CRN such that this bounded CRN already retains sufficient probability to refute the property under investigation. The effectiveness of this approach is demonstrated by analyzing rare-event properties on a number of biochemical systems.
PhageBox: An Open Source Digital Microfluidic Extension With Applications for Phage Discovery
Objective Recent advancements demonstrate the significant role of digital microfluidics in automating laboratory work with DNA and on-site viral testing. However, since commercially available instruments are limited to droplet manipulation, our work addresses the need for accelerated integration of other components, such as temperature control, that can expand the application domain. Methods We developed PhageBox—an accessible device that can be used as a biochip extension. At hardware level, PhageBox integrates temperature and electromagnetic control modules. At software level, PhageBox is controlled by embedded software containing a unique model for bio-protocol programming, and a graphical user interface for visual device feedback and operation. Results To evaluate PhageBox’s efficacy for biomedical applications, we performed functional testing. Similarly, we validated the temperature control using thermography, obtaining a range of ± 0.2 ∘C . The electromagnets produced a magnetic force of 15 milliTesla, demonstrating precise immobilization of magnetic beads. We show the potential of PhageBox for bacteriophage research through three initial protocols a universal framework for PCR, T7 bacteriophage restriction enzyme digestion, and concentrating ϕX174 RF genomic DNA. Conclusion Our work presents an open-source hardware and software extension for digital microfluidics devices. This extension integrates temperature and electromagnetic modules, demonstrating efficacy in biomedical applications and potential for bacteriophage research. Significance We developed PhageBox to be accessible the components are off-the-shelf at a low cost ( ≤ $ 200), and the hardware designs and software code are open-source. With the long aim of ensuring reproducibility and accelerating collaboration, we also provide a DIY-build document.
Evaluating the Contribution of Model Complexity in Predicting Robustness in Synthetic Genetic Circuits
The design–build–test–learn workflow is pivotal in synthetic biology as it seeks to broaden access to diverse levels of expertise and enhance circuit complexity through recent advancements in automation. The design of complex circuits depends on developing precise models and parameter values for predicting the circuit performance and noise resilience. However, obtaining characterized parameters under diverse experimental conditions is a significant challenge, often requiring substantial time, funding, and expertise. This work compares five computational models of three different genetic circuit implementations of the same logic function to evaluate their relative predictive capabilities. The primary focus is on determining whether simpler models can yield conclusions similar to those of more complex ones and whether certain models offer greater analytical benefits. These models explore the influence of noise, parametrization, and model complexity on predictions of synthetic circuit performance through simulation. The findings suggest that when developing a new circuit without characterized parts or an existing design, any model can effectively predict the optimal implementation by facilitating qualitative comparison of designs’ failure probabilities (e.g., higher or lower). However, when characterized parts are available and accurate quantitative differences in failure probabilities are desired, employing a more precise model with characterized parts becomes necessary, albeit requiring additional effort.
Ten simple rules for managing laboratory information
Author summary Information is the cornerstone of research, from experimental (meta)data and computational processes to complex inventories of reagents and equipment. These 10 simple rules discuss best practices for leveraging laboratory information management systems to transform this large information load into useful scientific findings.
Advancing reuse of genetic parts: progress and remaining challenges
Issues with data reuse have been recognized in synthetic biology and the broader scientific community. Policies and standards fall short as machine reasoning is not emphasised and enforcement is lacking. We discuss the progress, remaining challenges, and possible solutions.
Robustness and reproducibility of simple and complex synthetic logic circuit designs using a DBTL loop
Computational tools addressing various components of design–build–test–learn (DBTL) loops for the construction of synthetic genetic networks exist but do not generally cover the entire DBTL loop. This manuscript introduces an end-to-end sequence of tools that together form a DBTL loop called Design Assemble Round Trip (DART). DART provides rational selection and refinement of genetic parts to construct and test a circuit. Computational support for experimental process, metadata management, standardized data collection and reproducible data analysis is provided via the previously published Round Trip (RT) test–learn loop. The primary focus of this work is on the Design Assemble (DA) part of the tool chain, which improves on previous techniques by screening up to thousands of network topologies for robust performance using a novel robustness score derived from dynamical behavior based on circuit topology only. In addition, novel experimental support software is introduced for the assembly of genetic circuits. A complete design-through-analysis sequence is presented using several OR and NOR circuit designs, with and without structural redundancy, that are implemented in budding yeast. The execution of DART tested the predictions of the design tools, specifically with regard to robust and reproducible performance under different experimental conditions. The data analysis depended on a novel application of machine learning techniques to segment bimodal flow cytometry distributions. Evidence is presented that, in some cases, a more complex build may impart more robustness and reproducibility across experimental conditions.
Synthetic biology open language (SBOL) version 3.1.0
Synthetic biology builds upon genetics, molecular biology, and metabolic engineering by applying engineering principles to the design of biological systems. When designing a synthetic system, synthetic biologists need to exchange information about multiple types of molecules, the intended behavior of the system, and actual experimental measurements. The Synthetic Biology Open Language (SBOL) has been developed as a standard to support the specification and exchange of biological design information in synthetic biology, following an open community process involving both bench scientists and scientific modelers and software developers, across academia, industry, and other institutions. This document describes SBOL 3.1.0, which improves on version 3.0.0 by including a number of corrections and clarifications as well as several other updates and enhancements. First, this version includes a complete set of validation rules for checking whether documents are valid SBOL 3. Second, the best practices section has been moved to an online repository that allows for more rapid and interactive of sharing these conventions. Third, it includes updates based upon six community approved enhancement proposals. Two enhancement proposals are related to the representation of an object’s namespace. In particular, the Namespace class has been removed and replaced with a namespace property on each class. Another enhancement is the generalization of the CombinatorialDeriviation class to allow direct use of Features and Measures. Next, the Participation class now allow Interactions to be participants to describe higher-order interactions. Another change is the use of Sequence Ontology terms for Feature orientation. Finally, this version of SBOL has generalized from using Unique Reference Identifiers (URIs) to Internationalized Resource Identifiers (IRIs) to support international character sets.
SynBioSuite: A Tool for Improving the Workflow for Genetic Design and Modeling
Synthetic biology research has led to the development of many software tools for designing, constructing, editing, simulating, and sharing genetic parts and circuits. Among these tools are SBOLCanvas, iBioSim, and SynBioHub, which can be used in conjunction to create a genetic circuit design following the design–build–test–learn process. However, although automation works within these tools, most of these software tools are not integrated, and the process of transferring information between them is a very manual, error-prone process. To address this problem, this work automates some of these processes and presents SynBioSuite, a cloud-based tool that eliminates many of the drawbacks of the current approach by automating the setup and reception of results for simulating a designed genetic circuit via an application programming interface.
STAMINA in C++: Modernizing an Infinite-State Probabilistic Model Checker
Improving the scalability of probabilistic model checking (PMC) tools is crucial to the verification of real-world system designs. The Stamina infinite-state PMC tool achieves scalability by iteratively constructing a partial state space for an unbounded continuous-time Markov chain model, where a majority of the probability mass resides. It then performs time-bounded transient PMC. It can efficiently produce an accurate probability bound to the property under verification. We present a new software architecture design and the C++ implementation of the Stamina 2.0 algorithm, integrated with the Storm model checker. This open-source Stamina implementation offers a high degree of modularity and provides significant optimizations to the Stamina 2.0 algorithm. Performance improvements are demonstrated on multiple challenging benchmark examples, including hazard analysis of infinite-state combinational genetic circuits, over the previous Stamina implementation. Additionally, its design allows for future customizations and optimizations to the Stamina algorithm.
Specifications of standards in systems and synthetic biology: status and developments in 2022 and the COMBINE meeting 2022
This special issue of the Journal of Integrative Bioinformatics contains updated specifications of COMBINE standards in systems and synthetic biology. The 2022 special issue presents three updates to the standards: CellML 2.0.1, SBML Level 3 Package: Spatial Processes, Version 1, Release 1, and Synthetic Biology Open Language (SBOL) Version 3.1.0. This document can also be used to identify the latest specifications for all COMBINE standards. In addition, this editorial provides a brief overview of the COMBINE 2022 meeting in Berlin.
Investigating and Modeling the Factors That Affect Genetic Circuit Performance
Over the past 2 decades, synthetic biology has yielded ever more complex genetic circuits that are able to perform sophisticated functions in response to specific signals. Yet, genetic circuits are not immediately transferable to an outside-the-lab setting where their performance is highly compromised. We propose introducing a broader test step to the design–build–test–learn workflow to include factors that might contribute to unexpected genetic circuit performance. As a proof of concept, we have designed and evaluated a genetic circuit in various temperatures, inducer concentrations, nonsterilized soil exposure, and bacterial growth stages. We determined that the circuit’s performance is dramatically altered when these factors differ from the optimal lab conditions. We observed significant changes in the time for signal detection as well as signal intensity when the genetic circuit was tested under nonoptimal lab conditions. As a learning effort, we then proceeded to generate model predictions in untested conditions, which is currently lacking in synthetic biology application design. Furthermore, broader test and learn steps uncovered a negative correlation between the time it takes for a gate to turn ON and the bacterial growth phases. As the synthetic biology discipline transitions from proof-of-concept genetic programs to appropriate and safe application implementations, more emphasis on test and learn steps (i.e., characterizing parts and circuits for a broad range of conditions) will provide missing insights on genetic circuit behavior outside the lab.
Experimental Data Connector (XDC): Integrating the Capture of Experimental Data and Metadata Using Standard Formats and Digital Repositories
Accelerating the development of synthetic biology applications requires reproducible experimental findings. Different standards and repositories exist to exchange experimental data and metadata. However, the associated software tools often do not support a uniform data capture, encoding, and exchange of information. A connection between digital repositories is required to prevent siloing and loss of information. To this end, we developed the Experimental Data Connector (XDC). It captures experimental data and related metadata by encoding it in standard formats and storing the converted data in digital repositories. Experimental data is then uploaded to Flapjack and the metadata to SynBioHub in a consistent manner linking these repositories. This produces complete connected experimental data sets that are exchangeable. The information is captured using a single template Excel Workbook, which can be integrated into existing experimental workflow automation processes and semiautomated capture of results.
Experimental Data Connector (XDC): Integrating the Capture of Experimental Data and Metadata Using Standard Formats and Digital Repositories
Accelerating the development of synthetic biology applications requires reproducible experimental findings. Different standards and repositories exist to exchange experimental data and metadata. However, the associated software tools often do not support a uniform data capture, encoding, and exchange of information. A connection between digital repositories is required to prevent siloing and loss of information. To this end, we developed the Experimental Data Connector (XDC). It captures experimental data and related metadata by encoding it in standard formats and storing the converted data in digital repositories. Experimental data is then uploaded to Flapjack and the metadata to SynBioHub in a consistent manner linking these repositories. This produces complete connected experimental data sets that are exchangeable. The information is captured using a single template Excel Workbook, which can be integrated into existing experimental workflow automation processes and semiautomated capture of results.
Excel–SBOL Converter: Creating SBOL from Excel Templates and Vice Versa
Standards support synthetic biology research by enabling the exchange of component information. However, using formal representations, such as the Synthetic Biology Open Language (SBOL), typically requires either a thorough understanding of these standards or a suite of tools developed in concurrence with the ontologies. Since these tools may be a barrier for use by many practitioners, the Excel–SBOL Converter was developed to facilitate the use of SBOL and integration into existing workflows. The converter consists of two Python libraries: one that converts Excel templates to SBOL and another that converts SBOL to an Excel workbook. Both libraries can be used either directly or via a SynBioHub plugin.
Highly-automated, high-throughput replication of yeast-based logic circuit design assessments
We describe an experimental campaign that replicated the performance assessment of logic gates engineered into cells of Saccharomyces cerevisiae by Gander et al. Our experimental campaign used a novel high-throughput experimentation framework developed under Defense Advanced Research Projects Agency’s Synergistic Discovery and Design program: a remote robotic lab at Strateos executed a parameterized experimental protocol. Using this protocol and robotic execution, we generated two orders of magnitude more flow cytometry data than the original experiments. We discuss our results, which largely, but not completely, agree with the original report and make some remarks about lessons learned.Graphical Abstract
Investigating Genetic Circuit Failures
Synthetic biology is an engineering discipline in which biological components are assembled to form devices with user-defined functions. As a nascent discipline, genetic circuit design is reserved only for experienced researchers with an in-depth knowledge of biology. This work aims to alleviate some of these constraints by developing software to facilitate genetic circuit design and analysis so that more researchers can participate in this thriving discipline and help elucidate the causes of circuit failures. Firstly, an automatic dynamic model generator can be implemented to predict a circuit’s behavior between steady states and determine the amount of time needed to reach such steady states. Moreover, the analysis of the predicted dynamic behavior will help the designers understand the risks of applying specific input changes and decide whether the risk is critical for the designed systems’ intended purposes. Extrinsic and intrinsic noise can contribute to the observed output variability of a clonal population. Therefore, to account for a genetic circuits’ stochastic behavior, this work aims to develop stochastic modeling using extrinsic and intrinsic noise contributions that can help infer glitch probabilities and elucidate the causes of circuit failure. All the methodologies developed in this work will serve the overacting aim of redesigning genetic circuits to avoid circuit failures. Dynamic ODE modeling will predict glitching behavior and the time to reach said states; stochastic modeling will be used to predict glitch propensities; hazard-preserving transformations will be used to avoid solvable hazards. Facilitated dynamic modeling of genetic circuits would be an instrumental technique for synthetic biologists, especially if it can be accompanied by a circuit design automation tool, such as those proposed in this work. This would help automation in synthetic biology and provide a way to debug circuit designs before construction and compare predictions with experimental data once the synthesized circuit is implemented, saving time, effort, and money. This project aims to expand such capabilities to help researchers through the design process with the development of automated modeling, logic synthesis, hazard identification, and genetic circuit redesign.
BioSimulators: a central registry of simulation engines and services for recommending specific tools
Computational models have great potential to accelerate bioscience, bioengineering, and medicine. However, it remains challenging to reproduce and reuse simulations, in part, because the numerous formats and methods for simulating various subsystems and scales remain siloed by different software tools. For example, each tool must be executed through a distinct interface. To help investigators find and use simulation tools, we developed BioSimulators (https://biosimulators.org), a central registry of the capabilities of simulation tools and consistent Python, command-line and containerized interfaces to each version of each tool. The foundation of BioSimulators is standards, such as CellML, SBML, SED-ML and the COMBINE archive format, and validation tools for simulation projects and simulation tools that ensure these standards are used consistently. To help modelers find tools for particular projects, we have also used the registry to develop recommendation services. We anticipate that BioSimulators will help modelers exchange, reproduce, and combine simulations.
SynBioHub 3 - An Improved Synthetic Biology Repository
SynBioHub is a repository for storing, sharing, and finding modular genetic designs in synthetic biology. While valuable to the community, the original SynBioHub faces architectural limitations including coupled frontend-backend code, performance issues, and limited support for newer standards. This thesis presents SynBioHub3, a complete redesign using Java and Spring Boot that addresses these challenges through a strict separation of concerns based on the Model-View-Controller pattern. The new architecture creates a dedicated REST API interface, eliminates server-side rendering, and improves performance - notably in the download algorithm, which shows significant speed gains. SynBioHub3 implements core functionality including search, downloads, and authentication while maintaining backward compatibility with existing applications. This reimplementation provides a more maintainable, scalable foundation for synthetic biology data sharing that better supports FAIR principles and the community’s growing needs.
Genetic circuit design automation with Cello 2.0
Cells interact with their environment, communicate among themselves, track time and make decisions through functions controlled by natural regulatory genetic circuits consisting of interacting biological components. Synthetic programmable circuits used in therapeutics and other applications can be automatically designed by computer-aided tools. The Cello software designs the DNA sequences for programmable circuits based on a high-level software description and a library of characterized DNA parts representing Boolean logic gates. This process allows for design specification reuse, modular DNA part library curation and formalized circuit transformations based on experimental data. This protocol describes Cello 2.0, a freely available cross-platform software written in Java. Cello 2.0 enables flexible descriptions of the logic gates’ structure and their mathematical models representing dynamic behavior, new formal rules for describing the placement of gates in a genome, a new graphical user interface, support for Verilog 2005 syntax and a connection to the SynBioHub parts repository software environment. Collectively, these features expand Cello’s capabilities beyond Escherichia coli plasmids to new organisms and broader genetic contexts, including the genome. Designing circuits with Cello 2.0 produces an abstract Boolean network from a Verilog file, assigns biological parts to each node in the Boolean network, constructs a DNA sequence and generates highly structured and annotated sequence representations suitable for downstream processing and fabrication, respectively. The result is a sequence implementing the specified Boolean function in the organism and predictions of circuit performance. Depending on the size of the design space and users’ expertise, jobs may take minutes or hours to complete.
STAMINA 2.0: Improving Scalability of Infinite-State Stochastic Model Checking
Stochastic model checking (SMC) is a formal verification technique for the analysis of systems with probabilistic behavior. Scalability has been a major limiting factor for SMC tools to analyze real-world systems with large or infinite state spaces. The infinite-state Continuous-time Markov Chain (CTMC) model checker, STAMINA, tackles this problem by selectively exploring only a portion of a model’s state space, where a majority of the probability mass resides, to efficiently give an accurate probability bound to properties under verification. In this paper, we present two major improvements to STAMINA, namely, a method of calculating and distributing estimated state reachability probabilities that improves state space truncation efficiency and combination of the previous two CTMC analyses into one for generating the probability bound. Demonstration of the improvements on several benchmark examples, including hazard analysis of infinite-state combinational genetic circuits, yield significant savings in both run-time and state space size (and hence memory), compared to both the previous version of STAMINA and the infinite-state CTMC model checker INFAMY. The improved STAMINA demonstrates significant scalability to allow for the verification of complex real-world infinite-state systems.
Sequence-Based Searching for SynBioHub Using VSEARCH
The ability to search for a part by its sequence is crucial for a large repository of parts. Prior to this work, however, this was not possible on SynBioHub. Sequence-based search is now integrated into SynBioHub, allowing users to find a part by a sequence provided in plain text or a supported file format. This sequence-based search feature is accessible to users via SynBioHub’s web interface, or programmatically through its API. The core implementation of the tool uses VSEARCH, an open source, global alignment search tool, and it is integrated into SBOLExplorer, an open source distributed search engine used by SynBioHub. We present a new approach to scoring part similarity using SBOLExplorer, which takes into account both the popularity and percentage match of parts.
Round Trip: An Automated Pipeline for Experimental Design, Execution, and Analysis
Synthetic biology is a complex discipline that involves creating detailed, purpose-built designs from genetic parts. This process is often phrased as a Design-Build-Test-Learn loop, where iterative design improvements can be made, implemented, measured, and analyzed. Automation can potentially improve both the end-to-end duration of the process and the utility of data produced by the process. One of the most important considerations for the development of effective automation and quality data is a rigorous description of implicit knowledge encoded as a formal knowledge representation. The development of knowledge representation for the process poses a number of challenges, including developing effective human–machine interfaces, protecting against and repairing user error, providing flexibility for terminological mismatches, and supporting extensibility to new experimental types. We address these challenges with the DARPA SD2 Round Trip software architecture. The Round Trip is an open architecture that automates many of the key steps in the Test and Learn phases of a Design-Build-Test-Learn loop for high-throughput laboratory science. The primary contribution of the Round Trip is to assist with and otherwise automate metadata creation, curation, standardization, and linkage with experimental data. The Round Trip’s focus on metadata supports fast, automated, and replicable analysis of experiments as well as experimental situational awareness and experimental interpretability. We highlight the major software components and data representations that enable the Round Trip to speed up the design and analysis of experiments by 2 orders of magnitude over prior ad hoc methods. These contributions support a number of experimental protocols and experimental types, demonstrating the Round Trip’s breadth and extensibility. We describe both an illustrative use case using the Round Trip for an on-the-loop experimental campaign and overall contributions to reducing experimental analysis time and increasing data product volume in the SD2 program.
Promotion of Data Reuse in Synthetic Biology
Synthetic biology is a movement to standardize genetic engineering and make it more reproducible and accessible by using functional descriptions of desired circuits. Such descriptions can then be converted to genetic designs via genetic design automation tools. Subsequently, the genetic designs can be used to generate models for in silico experimentation using automatic model generators. Both of these technologies rely on access to libraries of genetic part information encoded in standard, machine-readable ways. The Synthetic Biology Open Language (SBOL) can be used together with SynBioHub (a genetic part repository) to encode and store the information. However, the use of SynBioHub for the storage and reuse of parts is still very limited. This is due to insufficient metadata (making it difficult to find parts or judge their usefulness) and the effort required to submit parts to the repository. This dissertation aims to decrease the barriers to part reuse and thus enable a more automated synthetic biology workflow. Hence, an integrated curation workflow is proposed based on the contributions of the dissertation. The contributions are: a proposed SBOL Data Content Standard, tools for working with genetic parts in spreadsheets, a framework to modularly extend the SynBioHub part repository, and the lessons learned from the analysis and curation of data from existing genetic data repositories.
Engineering genetic circuits: advancements in genetic design automation tools and standards for synthetic biology
Synthetic biology (SynBio) is a field at the intersection of biology and engineering. Inspired by engineering principles, researchers use defined parts to build functionally defined biological circuits. Genetic design automation (GDA) allows scientists to design, model, and analyze their genetic circuits in silico before building them in the lab, saving time, and resources in the process. Establishing SynBio’s future is dependent on GDA, since the computational approach opens the field to a broad, interdisciplinary community. However, challenges with part libraries, standards, and software tools are currently stalling progress in the field. This review first covers recent advancements in GDA, followed by an assessment of the challenges ahead, and a proposed automated genetic design workflow for the future.
Synthetic Biology Curation Tools (SYNBICT)
Much progress has been made in developing tools to generate component-based design representations of biological systems from standard libraries of parts. Most biological designs, however, are still specified at the sequence level. Consequently, there exists a need for a tool that can be used to automatically infer component-based design representations from sequences, particularly in cases when those sequences have minimal levels of annotation. Such a tool would assist computational synthetic biologists in bridging the gap between the outputs of sequence editors and the inputs to more sophisticated design tools, and it would facilitate their development of automated workflows for design curation and quality control. Accordingly, we introduce Synthetic Biology Curation Tools (SYNBICT), a Python tool suite for automation-assisted annotation, curation, and functional inference for genetic designs. We have validated SYNBICT by applying it to genetic designs in the DARPA Synergistic Discovery & Design (SD2) program and the International Genetically Engineered Machines (iGEM) 2018 distribution. Most notably, SYNBICT is more automated and parallelizable than manual design editors, and it can be applied to interpret existing designs instead of only generating new ones.
Curation Principles Derived from the Analysis of the SBOL iGEM Data Set
As an engineering endeavor, synthetic biology requires effective sharing of genetic design information that can be reused in the construction of new designs. While there are a number of large community repositories of design information, curation of this information has been limited. This in turn limits the ways in which design information can be put to use. The aim of this work was to improve this situation by creating a curated library of parts from the International Genetically Engineered Machines (iGEM) registry data set. To this end, an analysis of the Synthetic Biology Open Language (SBOL) version of the iGEM registry was carried out using four different approaches—simple statistics, SnapGene autoannotation, SYNBICT autoannotation, and expert analysis—the results of which are presented herein. Key challenges encountered include the use of free text, insufficient part provenance, part duplication, lack of part removal, and insufficient continuous curation. On the basis of these analyses, the focus has shifted from the creation of a curated iGEM part library to instead the extraction of a set of lessons, which are presented here. These lessons can be exploited to facilitate the creation and curation of other part libraries using a simpler and less labor intensive process.
Stochastic Hazard Analysis of Genetic Circuits in iBioSim and STAMINA
In synthetic biology, combinational circuits are used to program cells for various new applications like biosensors, drug delivery systems, and biofuels. Similar to asynchronous electronic circuits, some combinational genetic circuits may show unwanted switching variations (glitches) caused by multiple input changes. Depending on the biological circuit, glitches can cause irreversible effects and jeopardize the circuit’s functionality. This paper presents a stochastic analysis to predict glitch propensities for three implementations of a genetic circuit with known glitching behavior. The analysis uses STochastic Approximate Model-checker for INfinite-state Analysis (STAMINA), a tool for stochastic verification. The STAMINA results were validated by comparison to stochastic simulation in iBioSim resulting in further improvements of STAMINA. This paper demonstrates that stochastic verification can be utilized by genetic designers to evaluate design choices and input restrictions to achieve a desired reliability of operation.
Synthetic Biology Knowledge System
The Synthetic Biology Knowledge System (SBKS) is an instance of the SynBioHub repository that includes text and data information that has been mined from papers published in ACS Synthetic Biology. This paper describes the SBKS curation framework that is being developed to construct the knowledge stored in this repository. The text mining pipeline performs automatic annotation of the articles using natural language processing techniques to identify salient content such as key terms, relationships between terms, and main topics. The data mining pipeline performs automatic annotation of the sequences extracted from the supplemental documents with the genetic parts used in them. Together these two pipelines link genetic parts to papers describing the context in which they are used. Ultimately, SBKS will reduce the time necessary for synthetic biologists to find the information necessary to complete their designs.
Synthetic Biology Open Language Visual (SBOL Visual) Version 3.0
People who engineer biological organisms often find it useful to draw diagrams in order to communicate both the structure of the nucleic acid sequences that they are engineering and the functional relationships between sequence features and other molecular species. Some typical practices and conventions have begun to emerge for such diagrams. SBOL Visual aims to organize and systematize such conventions in order to produce a coherent language for expressing the structure and function of genetic designs. This document details version 3.0 of SBOL Visual, a new major revision of the standard. The major difference between SBOL Visual 3 and SBOL Visual 2 is that diagrams and glyphs are defined with respect to the SBOL 3 data model rather than the SBOL 2 data model. A byproduct of this change is that the use of dashed undirected lines for subsystem mappings has been removed, pending future determination on how to represent general SBOL 3 constraints; in the interim, this annotation can still be used as an annotation. Finally, deprecated material has been removed from collection of glyphs: the deprecated insulator'' glyph and macromolecule’’ alternative glyphs have been removed, as have the deprecated BioPAX alternatives to SBO terms.
Specifications of standards in systems and synthetic biology: status and developments in 2021
This special issue of the Journal of Integrative Bioinformatics contains updated specifications of COMBINE standards in systems and synthetic biology. The 2021 special issue presents four updates of standards: Synthetic Biology Open Language Visual Version 2.3, Synthetic Biology Open Language Visual Version 3.0, Simulation Experiment Description Markup Language Level 1 Version 4, and OMEX Metadata specification Version 1.2. This document can also be consulted to identify the latest specifications of all COMBINE standards.
iBioSim Server: A Tool for Improving the Workflow for Genetic Design and Modeling
With this work, a containerized API was created that is capable of automating the simulation of genetic circuits designed in SBOLCanvas using the working parts of the iBioSim tool. This API will help in the Design, Build, Test, Learn (DBTL) workflow for research in synthetic biology.
Comparison of Extrinsic and Intrinsic Noise Model Predictions for Genetic Circuit Failures
This work focuses on determining if there are differences in predicted circuit failure percentages for three different circuit layouts with identical expected functions, using stochastic analysis to simulate different noise sources. The results shed light on the difference between the intrinsic and extrinsic noise model predictions and if the differences in circuit layouts have any effect on glitch propensities. This, in turn, emphasizes the need to evaluate further the relative influence of intrinsic and extrinsic noise on a genetic circuit’s output to help designers predict circuit failures more accurately and, therefore, determine better design choices. Moreover, the percent failure predictions between different circuit layouts can help designers weigh different options of circuit topologies to determine which one is best suited for the intended purposes of the design.
A Comparison of Weighted Stochastic Simulation Methods
Rare events are of particular interest in biology because rare biochemical events may be catastrophic to a biological system. To estimate the probability of rare events, several weighted stochastic simulation methods have been developed. Unfortunately, the robustness of these methods is questionable. Here, an analysis of weighted stochastic simulation methods is presented. The methods considered here fail to accomplish the task of rare event simulation, in general, suggesting that new methods are necessary to adequately study rare biological events.
VisBOL2—Improving Web-Based Visualization for Synthetic Biology Designs
VisBOL is a web-based visualization tool used to depict genetic circuit designs. This tool depicts simple DNA circuits adequately, but it has become increasingly outdated as new versions of SBOL Visual were released. This paper introduces VisBOL2, a heavily redesigned version of VisBOL that makes a number of improvements to the original VisBOL, including proper functional interaction rendering, dynamic viewing, a more maintainable code base, and modularity that facilitates compatibility with other software tools. This modularity is demonstrated by incorporating VisBOL2 into a sequence visualization plugin for SynBioHub.
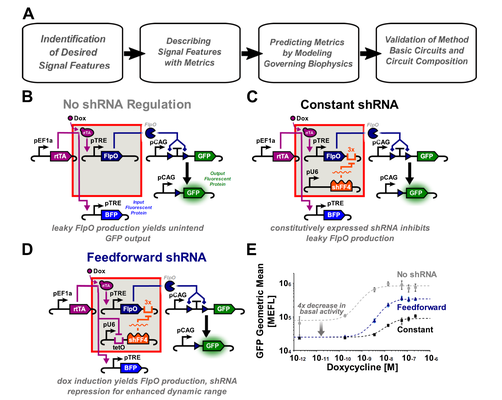
Quantitative Characterization of Recombinase-Based Digitizer Circuits Enables Predictable Amplification of Biological Signals
Many synthetic gene circuits are restricted to single-use applications or require iterative refinement for incorporation into complex systems. One example is the recombinase-based digitizer circuit, which has been used to improve weak or leaky biological signals. Here we present a workflow to quantitatively define digitizer performance and predict responses to different input signals. Using a combination of signal-to-noise ratio (SNR), area under a receiver operating characteristic curve (AUC), and fold change (FC), we evaluate three small-molecule inducible digitizer designs demonstrating FC up to 508x and SNR up to 3.77 dB. To study their behavior further and improve modularity, we develop a mixed phenotypic/mechanistic model capable of predicting digitizer configurations that amplify a synNotch cell-to-cell communication signal ($Δ$ SNR up to 2.8 dB). We hope the metrics and modeling approaches here will facilitate incorporation of these digitizers into other systems while providing an improved workflow for gene circuit characterization. Kiwimagi & Letendre et al. present a workflow to quantitatively define recombinase-based digitizer and predict responses to different input signals. With a mechanistic/phenotypic model that can predict circuit performance, they generate a synthetic cell-cell communication device that amplifies a synNotch output signal.
SBOLCanvas: A Visual Editor for Genetic Designs
SBOLCanvas is a web-based application that can create and edit genetic constructs using the SBOL data and visual standards. SBOLCanvas allows a user to create a genetic design visually and structurally from start to finish. It also allows users to incorporate existing SBOL data from a SynBioHub repository. By the nature of being a web-based application, SBOLCanvas is readily accessible and easy to use. A live version of the latest release can be found at https://sbolcanvas.org.
Synthetic Biology Open Language Visual (SBOL Visual) Version 2.3
People who are engineering biological organisms often find it useful to communicate in diagrams, both about the structure of the nucleic acid sequences that they are engineering and about the functional relationships between sequence features and other molecular species. Some typical practices and conventions have begun to emerge for such diagrams. The Synthetic Biology Open Language Visual (SBOL Visual) has been developed as a standard for organizing and systematizing such conventions in order to produce a coherent language for expressing the structure and function of genetic designs. This document details version 2.3 of SBOL Visual, which builds on the prior SBOL Visual 2.2 in several ways. First, the specification now includes higher-level interactions with interactions,'' such as an inducer molecule stimulating a repression interaction. Second, binding with a nucleic acid backbone can be shown by overlapping glyphs, as with other molecular complexes. Finally, a new unspecified interaction’’ glyph is added for visualizing interactions whose nature is unknown, the insulator'' glyph is deprecated in favor of a new inert DNA spacer’’ glyph, and the polypeptide region glyph is recommended for showing 2A sequences.
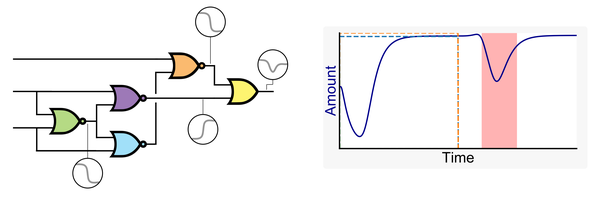
Genetic Circuit Dynamics: Hazard and Glitch Analysis
Multiple input changes can cause unwanted switching variations, or glitches, in the output of genetic combinational circuits. These glitches can have drastic effects if the output of the circuit causes irreversible changes within or with other cells such as a cascade of responses, apoptosis, or the release of a pharmaceutical in an off-target tissue. Therefore, avoiding unwanted variation of a circuit’s output can be crucial for the safe operation of a genetic circuit. This paper investigates what causes unwanted switching variations in combinational genetic circuits using hazard analysis and a new dynamic model generator. The analysis is done in previously built and modeled genetic circuits with known glitching behavior. The dynamic models generated not only predict the same steady states as previous models but can also predict the unwanted switching variations that have been observed experimentally. Multiple input changes may cause glitches due to propagation delays within the circuit. Modifying the circuit’s layout to alter these delays may change the likelihood of certain glitches, but it cannot eliminate the possibility that the glitch may occur. In other words, function hazards cannot be eliminated. Instead, they must be avoided by restricting the allowed input changes to the system. Logic hazards, on the other hand, can be avoided using hazard-free logic synthesis. This paper demonstrates this by showing how a circuit designed using a popular genetic design automation tool can be redesigned to eliminate logic hazards.
SBML Level 3: an extensible format for the exchange and reuse of biological models
Systems biology has experienced dramatic growth in the number, size, and complexity of computational models. To reproduce simulation results and reuse models, researchers must exchange unambiguous model descriptions. We review the latest edition of the Systems Biology Markup Language (SBML), a format designed for this purpose. A community of modelers and software authors developed SBML Level 3 over the past decade. Its modular form consists of a core suited to representing reaction-based models and packages that extend the core with features suited to other model types including constraint-based models, reaction-diffusion models, logical network models, and rule-based models. The format leverages two decades of SBML and a rich software ecosystem that transformed how systems biologists build and interact with models. More recently, the rise of multiscale models of whole cells and organs, and new data sources such as single-cell measurements and live imaging, has precipitated new ways of integrating data with models. We provide our perspectives on the challenges presented by these developments and how SBML Level 3 provides the foundation needed to support this evolution.
Systems Biology Markup Language (SBML) Level 3 Package: Distributions, Version 1, Release 1
Biological models often contain elements that have inexact numerical values, since they are based on values that are stochastic in nature or data that contains uncertainty. The Systems Biology Markup Language (SBML) Level 3 Core specification does not include an explicit mechanism to include inexact or stochastic values in a model, but it does provide a mechanism for SBML packages to extend the Core specification and add additional syntactic constructs. The SBML Distributions package for SBML Level 3 adds the necessary features to allow models to encode information about the distribution and uncertainty of values underlying a quantity.
The first 10 years of the international coordination network for standards in systems and synthetic biology (COMBINE)
This paper presents a report on outcomes of the 10th Computational Modeling in Biology Network (COMBINE) meeting that was held in Heidelberg, Germany, in July of 2019. The annual event brings together researchers, biocurators and software engineers to present recent results and discuss future work in the area of standards for systems and synthetic biology. The COMBINE initiative coordinates the development of various community standards and formats for computational models in the life sciences. Over the past 10 years, COMBINE has brought together standard communities that have further developed and harmonized their standards for better interoperability of models and data. COMBINE 2019 was co-located with a stakeholder workshop of the European EU-STANDS4PM initiative that aims at harmonized data and model standardization for in silico models in the field of personalized medicine, as well as with the FAIRDOM PALs meeting to discuss findable, accessible, interoperable and reusable (FAIR) data sharing. This report briefly describes the work discussed in invited and contributed talks as well as during breakout sessions. It also highlights recent advancements in data, model, and annotation standardization efforts. Finally, this report concludes with some challenges and opportunities that this community will face during the next 10 years.
Synthetic biology open language (SBOL) version 3.0.0
Synthetic biology builds upon genetics, molecular biology, and metabolic engineering by applying engineering principles to the design of biological systems. When designing a synthetic system, synthetic biologists need to exchange information about multiple types of molecules, the intended behavior of the system, and actual experimental measurements. The Synthetic Biology Open Language (SBOL) has been developed as a standard to support the specification and exchange of biological design information in synthetic biology, following an open community process involving both wet bench scientists and dry scientific modelers and software developers, across academia, industry, and other institutions. This document describes SBOL 3.0.0, which condenses and simplifies previous versions of SBOL based on experiences in deployment across a variety of scientific and industrial settings. In particular, SBOL 3.0.0, (1) separates sequence features from part/sub-part relationships, (2) renames Component Definition/Component to Component/Sub-Component, (3) merges Component and Module classes, (4) ensures consistency between data model and ontology terms, (5) extends the means to define and reference Sub-Components, (6) refines requirements on object URIs, (7) enables graph-based serialization, (8) moves Systems Biology Ontology (SBO) for Component types, (9) makes all sequence associations explicit, (10) makes interfaces explicit, (11) generalizes Sequence Constraints into a general structural Constraint class, and (12) expands the set of allowed constraints.
Synthetic biology open language visual (SBOL visual) version 2.2
People who are engineering biological organisms often find it useful to communicate in diagrams, both about the structure of the nucleic acid sequences that they are engineering and about the functional relationships between sequence features and other molecular species. Some typical practices and conventions have begun to emerge for such diagrams. The Synthetic Biology Open Language Visual (SBOL Visual) has been developed as a standard for organizing and systematizing such conventions in order to produce a coherent language for expressing the structure and function of genetic designs. This document details version 2.2 of SBOL Visual, which builds on the prior SBOL Visual 2.1 in several ways. First, the grounding of molecular species glyphs is changed from BioPAX to SBO, aligning with the use of SBO terms for interaction glyphs. Second, new glyphs are added for proteins, introns, and polypeptide regions (e. g., protein domains), the prior recommended macromolecule glyph is deprecated in favor of its alternative, and small polygons are introduced as alternative glyphs for simple chemicals.
Extending SynBioHub’s Functionality with Plugins
SynBioHub is a repository for synthetic genetic designs represented in the Synthetic Biology Open Language (SBOL). To integrate SynBioHub into more synthetic biology workflows, its data processing capabilities need to be expanded. To this end, a plugin interface has been developed. Plugins can be developed for data submission, visualization, and download. This framework was tested by the development of three example plugins, one of each type as follows: one allowing the submission of SnapGene files, one visualizing the course of different genetic parts, and one preparing plasmid maps for download.
The Synthetic Biology Open Language (SBOL) Version 3: Simplified Data Exchange for Bioengineering
The Synthetic Biology Open Language (SBOL) is a community-developed data standard that allows knowledge about biological designs to be captured using a machine-tractable, ontology-backed representation that is built using Semantic Web technologies. While early versions of SBOL focused only on the description of DNA-based components and their sub-components, SBOL can now be used to represent knowledge across multiple scales and throughout the entire synthetic biology workflow, from the specification of a single molecule or DNA fragment through to multicellular systems containing multiple interacting genetic circuits. The third major iteration of the SBOL standard, SBOL3, is an effort to streamline and simplify the underlying data model with a focus on real-world applications, based on experience from the deployment of SBOL in a variety of scientific and industrial settings. Here, we introduce the SBOL3 specification both in comparison to previous versions of SBOL and through practical examples of its use.
Improving Authentication and Authorization on SynBioHub
Synthetic Biology is an emerging discipline which uses engineering principles to shape biological behavior. The Synthetic Biology Open Language (SBOL) is a standard for describing biological constructs which enables engineering workflows that previous formats, such as GenBank and FASTA, could not. SynBioHub is an online repository for storing and sharing genetic designs. It uses the SBOL standard and an RDF triplestore to store designs, as well as supporting file attachment and external links. Several research efforts in synthetic biology have adopted SynBioHub and SBOL. These research efforts have revealed key areas for improvement in SynBioHub. Improving user sharing and permissioning is a primary target for improvement. The existing system has basic support for sharing with different privilege levels. Unfortunately, its architecture makes it difficult to extend and improve. Due to this difficulty, many features which would make SynBioHub more collaborative have not been implemented. This work aims to make synthetic biology more collaborative by providing a better foundation for experimentation and innovation in user sharing and permissioning. The existing authentication and authorization (auth) system is not centralized; it mixes concerns between page rendering and permissions management. The new system separates auth into its own software layer, separate entirely from page rendering. This new layer is itself split into separate authentication and authorization steps. New feature development and refinement will be made easier by the strong separations between the different components of SynBioHub.
Automated Generation of Dynamic Models for Genetic Regulatory Networks
Synthetic biology is an engineering discipline in which biological components are assembled to form devices with user-defined functions. As in any engineering discipline, modeling is a big part of the design process, since it helps to predict, control, and debug systems in an efficient manner. Systems biology has always been concerned with dynamic models, and a recent increase in high-throughput of experimental data has made it essential to develop dynamic models that can be used for an iterative learning process in a design/build/test workflow. In this thesis work, an automated model generator is created to automatically generate dynamic models for genetic regulatory networks, implemented in the genetic design automation tool, iBioSim. This automated model generator uses parameters stored at an online parts repository and encodes the mathematical models it generates using Systems Biology Markup Language. The automated model generator is then used to model and simulate genetic circuits created with the design environment referred to as Cello. The simulation of the mathematical models produces a dynamical response prediction of each of the circuits, which is unavailable with steady-state modeling. Some of these dynamical responses present unexpected behavior. Using the dynamic models generated with the automatic model generator of this work, an analysis of the predicted behaviors yielded insight into the underlying biology phenomena that cause the observed glitching behavior of these circuits. The last chapter of this thesis is focused mainly on future enhancements to the automated model generator of this work to produce more accurate and precise models not only for genetic regulatory networks in textitEscherichia coli, but any organism where parametrization exists as proposed in this thesis work. It also explores different analysis that could be implemented into the automated model generator of this work, in order to expand the assessment done on genetic circuits.
Asynchronous Genetic Design
Synthetic biology is applying engineering concepts to biological processes to enable genetic circuit designs, among other applications. As more biological parts are being discovered, it is vital to have an automated procedure to allow complex circuit designs to be built. Technology mapping is a set of procedures that maps biological components to a design specification. Current technology mapping frameworks for genetic circuits are used to design combinational circuits. This dissertation illustrates the process of building an automated workflow for a technology mapping framework to design synchronous sequential genetic circuits. An automated process to create a library of gates for logic and memory circuits is described to construct gates from DNA parts retrieved from a standardize data repository. Genetic constraints address what parts can be mapped to the design specification when the gates and designs are constructed. The proposed automaton workflow begins with a specification provided in a formal design language, such as Verilog. The input design specification is converted into a genetic regulatory network represented using the Synthetic Biology Open Language (SBOL). The network is decomposed into base functions (NOR gates, inverters, and genetic toggle switches) and matching and covering algorithms are performed to produce the output design. The output design is converted to the Systems Biology Markup Language (SBML) data format for testing and simulation. The outcome of this work provides the synthetic biology community insights on how asynchronous sequential circuit designs can be built through an automated procedure to perform technology mapping from libraries composed of logic gates and memory circuits.
Specifying Combinatorial Designs with the Synthetic Biology Open Language (SBOL)
As improvements in DNA synthesis technology and assembly methods make combinatorial assembly of genetic constructs increasingly accessible, methods for representing genetic constructs likewise need to improve to handle the exponential growth of combinatorial design space. To this end, we present a community accepted extension of the SBOL data standard that allows for the efficient and flexible encoding of combinatorial designs. This extension includes data structures for representing genetic designs with “variable” components that can be implemented by choosing one of many linked designs for existing genetic parts or constructs. We demonstrate the representational power of the SBOL combinatorial design extension through case studies on metabolic pathway design and genetic circuit design, and we report the expansion of the SBOLDesigner software tool to support users in creating and modifying combinatorial designs in SBOL.
SBOL-OWL: An Ontological Approach for Formal and Semantic Representation of Synthetic Biology Information
Standard representation of data is key for the reproducibility of designs in synthetic biology. The Synthetic Biology Open Language (SBOL) has already emerged as a data standard to represent information about genetic circuits, and it is based on capturing data using graphs. The language provides the syntax using a free text document that is accessible to humans only. This paper describes SBOL-OWL, an ontology for a machine understandable definition of SBOL. This ontology acts as a semantic layer for genetic circuit designs. As a result, computational tools can understand the meaning of design entities in addition to parsing structured SBOL data. SBOL-OWL not only describes how genetic circuits can be constructed computationally, it also facilitates the use of several existing Semantic Web tools for synthetic biology. This paper demonstrates some of these features, for example, to validate designs and check for inconsistencies. Through the use of SBOL-OWL, queries can be simplified and become more intuitive. Moreover, existing reasoners can be used to infer information about genetic circuit designs that cannot be directly retrieved using existing querying mechanisms. This ontological representation of the SBOL standard provides a new perspective to the verification, representation, and querying of information about genetic circuits and is important to incorporate complex design information via the integration of biological ontologies.
pySBOL: A Python Package for Genetic Design Automation and Standardization
This paper presents pySBOL, a software library for computer-aided design of synthetic biological systems in the Python scripting language. This library provides an easy-to-use, object-oriented, application programming interface (API) with low barrier of entry for synthetic biology application developers. The pySBOL library enables reuse of genetic parts and designs through standardized data exchange with biological parts repositories and software tools that communicate using the Synthetic Biology Open Language (SBOL). In addition, pySBOL supports data management of design-build-test-learn workflows for individual laboratories as well as large, distributed teams of synthetic biologists. PySBOL also lets users add custom data to SBOL files to support the specific data requirements of their research. This extensibility helps users integrate software tool chains and develop workflows for new applications. These features and others make the pySBOL library a valuable tool for supporting engineering practices in synthetic biology. Documentation and installation instructions can be found at pysbol2.readthedocs.io.
iBioSim 3: A Tool for Model-Based Genetic Circuit Design
The iBioSim tool has been developed to facilitate the design of genetic circuits via a model-based design strategy. This paper illustrates the new features incorporated into the tool for DNA circuit design, design analysis, and design synthesis, all of which can be used in a workflow for the systematic construction of new genetic circuits.
A Computational Workflow for the Automated Generation of Models of Genetic Designs
Computational models are essential to engineer predictable biological systems and to scale up this process for complex systems. Computational modeling often requires expert knowledge and data to build models. Clearly, manual creation of models is not scalable for large designs. Despite several automated model construction approaches, computational methodologies to bridge knowledge in design repositories and the process of creating computational models have still not been established. This paper describes a workflow for automatic generation of computational models of genetic circuits from data stored in design repositories using existing standards. This workflow leverages the software tool SBOLDesigner to build structural models that are then enriched by the Virtual Parts Repository API using Systems Biology Open Language (SBOL) data fetched from the SynBioHub design repository. The iBioSim software tool is then utilized to convert this SBOL description into a computational model encoded using the Systems Biology Markup Language (SBML). Finally, this SBML model can be simulated using a variety of methods. This workflow provides synthetic biologists with easy to use tools to create predictable biological systems, hiding away the complexity of building computational models. This approach can further be incorporated into other computational workflows for design automation.
Specifications of Standards in Systems and Synthetic Biology: Status and Developments in 2019
This special issue of the Journal of Integrative Bioinformatics presents an overview of COMBINE standards and their latest specifications. The standards cover representation formats for computational modeling in synthetic and systems biology and include BioPAX, CellML, NeuroML, SBML, SBGN, SBOL and SED-ML. The articles in this issue contain updated specifications of SBGN Process Description Level 1 Version 2, SBML Level 3 Core Version 2 Release 2, SBOL Version 2.3.0, and SBOL Visual Version 2.1.
STAMINA: STochastic Approximate Model-Checker for INfinite-State Analysis
Stochastic model checking is a technique for analyzing systems that possess probabilistic characteristics. However, its scalability is limited as probabilistic models of real-world applications typically have very large or infinite state space. This paper presents a new infinite state CTMC model checker, STAMINA, with improved scalability. It uses a novel state space approximation method to reduce large and possibly infinite state CTMC models to finite state representations that are amenable to existing stochastic model checkers. It is integrated with a new property-guided state expansion approach that improves the analysis accuracy. Demonstration of the tool on several benchmark examples shows promising results in terms of analysis efficiency and accuracy compared with a state-of-the-art CTMC model checker that deploys a similar approximation method.
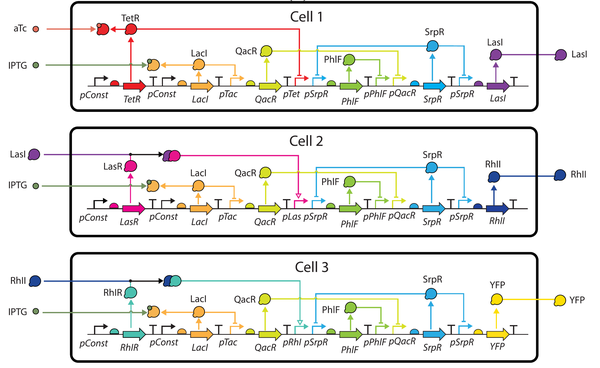
Design of Asynchronous Genetic Circuits
Most digital electronic circuits utilize a timing reference to synchronize the progression of signals and enable sequential memory elements. These designs may not be realizable in biological substrates due to the lack of a reliable high-frequency clock signal. Asynchronous designs eliminate the need for a clock with data encodings and request/acknowledge handshake protocols. This paper proposes a workflow to automate the design of asynchronous genetic circuits. This workflow extends genetic design tools by leveraging asynchronous logic design methods customized for this technology. This workflow is demonstrated on a genetic sensor that uses filtering and cellular communication to improve its reliability.
The Systems Biology Markup Language (SBML): Language Specification for Level 3 Version 2 Core Release 2
Computational models can help researchers to interpret data, understand biological functions, and make quantitative predictions. The Systems Biology Markup Language (SBML) is a file format for representing computational models in a declarative form that different software systems can exchange. SBML is oriented towards describing biological processes of the sort common in research on a number of topics, including metabolic pathways, cell signaling pathways, and many others. By supporting SBML as an input/output format, different tools can all operate on an identical representation of a model, removing opportunities for translation errors and assuring a common starting point for analyses and simulations. This document provides the specification for Release 2 of Version 2 of SBML Level 3 Core. The specification defines the data structures prescribed by SBML as well as their encoding in XML, the eXtensible Markup Language. Release 2 corrects some errors and clarifies some ambiguities discovered in Release 1. This specification also defines validation rules that determine the validity of an SBML document, and provides many examples of models in SBML form. Other materials and software are available from the SBML project website at http://sbml.org/ .
Synthetic Biology Open Language Visual (SBOL Visual) Version 2.1
People who are engineering biological organisms often find it useful to communicate in diagrams, both about the structure of the nucleic acid sequences that they are engineering and about the functional relationships between sequence features and other molecular species . Some typical practices and conventions have begun to emerge for such diagrams. The Synthetic Biology Open Language Visual (SBOL Visual) has been developed as a standard for organizing and systematizing such conventions in order to produce a coherent language for expressing the structure and function of genetic designs. This document details version 2.1 of SBOL Visual, which builds on the prior SBOL Visual 2.0 standard by expanding diagram syntax to include methods for showing modular structure and mappings between elements of a system, interactions arrows that can split or join (with the glyph at the split or join indicating either superposition or a chemical process), and adding new glyphs for indicating genomic context (e.g., integration into a plasmid or genome) and for stop codons.
Synthetic Biology Open Language (SBOL) Version 2.3
Synthetic biology builds upon the techniques and successes of genetics, molecular biology, and metabolic engineering by applying engineering principles to the design of biological systems. The field still faces substantial challenges, including long development times, high rates of failure, and poor reproducibility. One method to ameliorate these problems is to improve the exchange of information about designed systems between laboratories. The synthetic biology open language (SBOL) has been developed as a standard to support the specification and exchange of biological design information in synthetic biology, filling a need not satisfied by other pre-existing standards. This document details version 2.3.0 of SBOL, which builds upon version 2.2.0 published in last year’s JIB Standards in Systems Biology special issue. In particular, SBOL 2.3.0 includes means of succinctly representing sequence modifications, such as insertion, deletion, and replacement, an extension to support organization and attachment of experimental data derived from designs, and an extension for describing numerical parameters of design elements. The new version also includes specifying types of synthetic biology activities, unambiguous locations for sequences with multiple encodings, refinement of a number of validation rules, improved figures and examples, and clarification on a number of issues related to the use of external ontology terms.
Software Compliance Testing for Workflows Using the Synthetic Biology Open Language
Data standards are integral for interoperability between software applications, since they provide guidelines for how data can be meaningfully exchanged and in a uniform manner. While standards provide a bridge for applications to share and translate data, they do not guarantee that applications are compatible to perform a data exchange or that any translated data is legal and valid. As such, data passed from pairing applications must be validated to ensure that the data was not transformed or lost in the process of exchanging information. Ideally we would want an exchange between tools that is automatically successful; however, the data translated might not be legal or valid any longer. Therefore, data exchanges between applications need to be evaluated under conditions to ensure that compliance with the standard is met. The proposed research is to develop a compliance methodology that tests compliance of applications against the Synthetic Biology Open Language (SBOL) standard. This research aims to provide a robust test suite, a TestRunner tool implementing the compliance strategy, and a demonstration of the created methodology.
Scalable and Reproducible Modeling and Simulation for Heterogeneous Populations
Advancements in the systems and synthetic biology fields have proved that biology can be engineered. The development of computer-aided design (CAD) tools has contributed to advancements in these fields. Mathematical modeling and simulation methods are important assets of CAD tools that are frequently applied to the systems and synthetic biology fields. Modeling and simulation methods are used to understand or predict the behavior of a biological system being studied. However, many modeling efforts in those fields face a reproducibility problem, where many published models are not reproducible. In order to address such issue, standards have been created for the representation of biological models. A major advantage of standards is that they enable model reuse and sharing. The leading standard representation of biological systems is the Systems Biology Markup Language (SBML). The SBML standard is used to describe how biological processes affect and modify biological entities in a system. Such standard has been widely used to describe biochemical networks, cell signaling path, and gene regulation, among others. Unfortunately, not all models use SBML since there are many biological systems that SBML is incapable of representing efficiently, such as heterogeneous cellular populations. This dissertation explores extensions to SBML for the efficient representation of large heterogeneous cellular populations and simulation methods that can simulate such complex models efficiently. Since cellular populations are inherently hierarchical, this dissertation proposes an efficient simulator for hierarchical SBML models. Since the hierarchical structure is preserved in the proposed simulator, the hierarchical simulator is a perfect fit for handling hybrid models. However, no one has explored the coupling of different modeling formalisms within the same SBML model. Hence, this dissertation proposes a methodology that can be used to describe hybrid models. Such methodology is demonstrated by using dynamic flux balance analysis (DFBA) models as examples and such models can be successfully exchanged between tools. This dissertation also discusses extensions to the SBML data model to support regular structures in the form of arrays. Arrays is well-suited for population models since population models use large regular structures. Another application of arrays is microsimulation of disease models, where a population of individuals with unique characteristics need to be model. With the proposed arrays extension, simulators need to scale in order to handle the increase complexity that the arrays extension introduces. Hence, this dissertation also proposes an efficient simulation method that takes advantages of arrays.
SBOLExplorer: Data Infrastructure and Data Mining for Genetic Design Repositories
Biology is a very noisy field. Experiments are difficult to reproduce, the mechanisms behind life are not well understood, and data that we do obtain is difficult to make sense of. Much like traditional engineering fields where engineers draw from a library of reusable parts for their designs, experimental and synthetic biologists have designed biological circuits by drawing from a library of genetic constructs. However, these so-called genetic parts are poorly understood and are therefore limited in their usefulness. Additionally, there are hundreds of thousands of parts and sequences that have been either created or discovered. For my thesis, I filter through this biological noise to provide genetic circuit designers a powerful way to search for and access the genetic parts that are useful to them. This thesis is focused on creating SBOLExplorer, a system that is used to provide intuitive search within the SynBioHub genetic design repository. SynBioHub integrates genetic construct data from various sources and transforms and stores this data in a standardized data model. By tackling the intricate data mining and data infrastructure problems associated with large-scale semi-structured and noisy data, the search, transformation, and storage of data in genetic design repositories can be enhanced. In particular, this thesis focuses on improving the usability of genetic part repositories’ search capabilities. By clustering SynBioHub’s genetic parts into many derived collections, duplicate parts are merged. From there, a graph analysis algorithm is used to rank collections of parts by popularity and usefulness. Finally, data infrastructure challenges relating to indexing, storing, serving, and distributed search are solved. The end goal of SBOLExplorer is to integrate these findings into SynBioHub and other genetic design repositories’ data representation, search functionality, and data infrastructure.
sboljs: Bringing the Synthetic Biology Open Language to the Web Browser
The Synthetic Biology Open Language (SBOL) is a data standard for the representation of engineered biological systems. SBOL is implemented in the form of software libraries which can be used to add SBOL support to both new and existing software tools. While existing libraries allow for software to be developed that runs on a server or is installed locally, they lack the capability to create SBOL software that runs directly in a Web browser. Here, we address this issue by presenting sboljs, a JavaScript software library for SBOL that is capable of being used both on the server and in the Web browser.
Approximation techniques for stochastic analysis of biological systems
This book presents outstanding contributions in an exciting, new and multidisciplinary research area: the application of formal, automated reasoning techniques to analyse complex models in systems biology and systems medicine. Automated reasoning is a field of computer science devoted to the development of algorithms that yield trustworthy answers, providing a basis of sound logical reasoning. For example, in the semiconductor industry formal verification is instrumental to ensuring that chip designs are free of defects (or "bugs"). Over the past 15 years, systems biology and systems medicine have been introduced in an attempt to understand the enormous complexity of life from a computational point of view. This has generated a wealth of new knowledge in the form of computational models, whose staggering complexity makes manual analysis methods infeasible. Sound, trusted, and automated means of analysing the models are thus required in order to be able to trust their conclusions. Above all, this is crucial to engineering safe biomedical devices and to reducing our reliance on wet-lab experiments and clinical trials, which will in turn produce lower economic and societal costs. Some examples of the questions addressed here include: Can we automatically adjust medications for patients with multiple chronic conditions? Can we verify that an artificial pancreas system delivers insulin in a way that ensures Type 1 diabetic patients never suffer from hyperglycaemia or hypoglycaemia? And lastly, can we predict what kind of mutations a cancer cell is likely to undergo? This book brings together leading researchers from a number of highly interdisciplinary areas, including: - Parameter inference from time series - Model selection - Network structure identification - Machine learning - Systems medicine - Hypothesis generation from experimental data - Systems biology, systems medicine, and digital pathology - Verification of biomedical devices "This book presents a comprehensive spectrum of model-focused analysis techniques for biological systems …an essential resource for tracking the developments of a fast moving field that promises to revolutionize biology and medicine by the automated analysis of models and data." Prof Luca Cardelli FRS, University of Oxford
Synthetic Biology Open Language (SBOL) Version 2.2.0
Synthetic biology builds upon the techniques and successes of genetics, molecular biology, and metabolic engineering by applying engineering principles to the design of biological systems. The field still faces substantial challenges, including long development times, high rates of failure, and poor reproducibility. One method to ameliorate these problems would be to improve the exchange of information about designed systems between laboratories. The synthetic biology open language (SBOL) has been developed as a standard to support the specification and exchange of biological design information in synthetic biology, filling a need not satisfied by other pre-existing standards. This document details version 2.2.0 of SBOL that builds upon version 2.1.0 published in last year’s JIB special issue. In particular, SBOL 2.2.0 includes improved description and validation rules for genetic design provenance, an extension to support combinatorial genetic designs, a new class to add non-SBOL data as attachments, a new class for genetic design implementations, and a description of a methodology to describe the entire design-build-test-learn cycle within the SBOL data model.
SBOLDesigner: A Hierarchical Genetic Design Editor
Synthetic biology, as a field of research, applies electrical engineering, systems biology, and bioinformatics to genetic circuit design. Software tools are leveraged to provide rapid iteration through the design space, and data standards are used to encode and characterize complicated genetic circuit designs. Specifically, genetic design automation workflows centered around standards, abstraction, and decoupling are utilized to help experimental biologists accomplish their goals. Unfortunately, the software tools that support this workflow are lacking in some critical features such as combinatorial design and support for an extended range of glyphs. These inadequacies hinder the adoption of data standards, and therefore hurt the reproducibility of experiments and results. Necessary details of experiments are not recorded, and the resulting conclusions are therefore not trusted. SBOLDesigner, a sequence-based computer aided design tool, addresses these issues. This thesis will focus on SBOLDesigner’s implementation of combinatorial design and the SBOL Visual 2 standard using the SBOL 2 data model. Using SBOLDesigner, experimental biologists are able to visualize their genetic circuits unambiguously and express the full state of their design robustly. This results in higher productivity when designing genetic circuits, more comprehensive circuit descriptions, and most importantly, enhanced reproducibility in the field of synthetic biology.
Specifications of Standards in Systems and Synthetic Biology: Status and Developments in 2017
Standards are essential to the advancement of Systems and Synthetic Biology. COMBINE provides a formal body and a centralised platform to help develop and disseminate relevant standards and related resources. The regular special issue of the Journal of Integrative Bioinformatics aims to support the exchange, distribution and archiving of these standards by providing unified, easily citable access. This paper provides an overview of existing COMBINE standards and presents developments of the last year.
Synthetic Biology Open Language Visual (SBOL Visual) Version 2.0
People who are engineering biological organisms often find it useful to communicate in diagrams, both about the structure of the nucleic acid sequences that they are engineering and about the functional relationships between sequence features and other molecular species. Some typical practices and conventions have begun to emerge for such diagrams. The Synthetic Biology Open Language Visual (SBOL Visual) has been developed as a standard for organizing and systematizing such conventions in order to produce a coherent language for expressing the structure and function of genetic designs. This document details version 2.0 of SBOL Visual, which builds on the prior SBOL Visual 1.0 standard by expanding diagram syntax to include functional interactions and molecular species, making the relationship between diagrams and the SBOL data model explicit, supporting families of symbol variants, clarifying a number of requirements and best practices, and significantly expanding the collection of diagram glyphs.
The Systems Biology Markup Language (SBML): Language Specification for Level 3 Version 2 Core
Computational models can help researchers to interpret data, understand biological functions, and make quantitative predictions. The Systems Biology Markup Language (SBML) is a file format for representing computational models in a declarative form that different software systems can exchange. SBML is oriented towards describing biological processes of the sort common in research on a number of topics, including metabolic pathways, cell signaling pathways, and many others. By supporting SBML as an input/output format, different tools can all operate on an identical representation of a model, removing opportunities for translation errors and assuring a common starting point for analyses and simulations. This document provides the specification for Version 2 of SBML Level 3 Core. The specification defines the data structures prescribed by SBML, their encoding in XML (the eXtensible Markup Language), validation rules that determine the validity of an SBML document, and examples of models in SBML form. The design of Version 2 differs from Version 1 principally in allowing new MathML constructs, making more child elements optional, and adding identifiers to all SBML elements instead of only selected elements. Other materials and software are available from the SBML project website at http://sbml.org/.
SynBioHub: A Standards-Enabled Design Repository for Synthetic Biology
The SynBioHub repository (https://synbiohub.org) is an open-source software project that facilitates the sharing of information about engineered biological systems. SynBioHub provides computational access for software and data integration, and a graphical user interface that enables users to search for and share designs in a Web browser. By connecting to relevant repositories (e.g., the iGEM repository, JBEI ICE, and other instances of SynBioHub), the software allows users to browse, upload, and download data in various standard formats, regardless of their location or representation. SynBioHub also provides a central reference point for other resources to link to, delivering design information in a standardized format using the Synthetic Biology Open Language (SBOL). The adoption and use of SynBioHub, a community-driven effort, has the potential to overcome the reproducibility challenge across laboratories by helping to address the current lack of information about published designs.
A brief history of COMBINE
Standards for data exchange are critical to the development of any field. They enable researchers and practitioners to transport information reliably, to apply a variety of tools to their problems, and to reproduce scientific results. Over the past two decades, a range of standards have been developed to facilitate the exchange and reuse of information in the domain of representation and modeling of biological systems. These standards are complementary, so the interactions between their developers increased over time. By the end of the last decade, the community of researchers decided that more interoperability is required between the standards, and that common development is needed to make better use of effort, time, and money devoted to this activity. The COmputational MOdeling in Biology NEtwork (COMBINE) was created to enable the sharing of resources, tools, and other infrastructure. This paper provides a brief history of this endeavor and the challenges that remain.
Software Compliance Testing for the Synthetic Biology Open Language
Data Standards provide a way of expressing data in a uniform manner. In addition to standardizing a format for encoding data, data standards allow for data to be exchanged easily and meaningfully. Standards, commonly, enable applications to easily communicate and pass data to one another; however, this seamless communication between applications is impossible if applications rely on different data standards that encode data differently. This thesis proposes a workflow methodology for best-effort automatic conversion or translation of meta-data from one data standard to another while minimizing the loss of data. The objective of the methodology is to validate the conversion and determine the compatibility between two tools and their underlying data standards. The standard-enabled workflow and methodology created should analyze a given workflow of tools to see if data is lost within the workflow and ensure that the data is still compliant with a standard as the data flows through various tools. To determine how well the methodology works, Synthetic Biology tools are evaluated to see valid connections can be made with other tools while maintaining compliance within the data standard supported by the tool.
SBOLDesigner 2: An Intuitive Tool for Structural Genetic Design
As the Synthetic Biology Open Language (SBOL) data and visual standards gain acceptance for describing genetic designs in a detailed and reproducible way, there is an increasing need for an intuitive sequence editor tool that biologists can use that supports these standards. This paper describes SBOLDesigner 2, a genetic design automation (GDA) tool that natively supports both the SBOL data model (Version 2) and SBOL Visual (Version 1). This software is enabled to fetch and store parts and designs from SBOL repositories, such as SynBioHub. It can also import and export data about parts and designs in FASTA, GenBank, and SBOL 1 data format. Finally, it possesses a simple and intuitive user interface. This paper describes the design process using SBOLDesigner 2, highlighting new features over the earlier prototype versions. SBOLDesigner 2 is released freely and open source under the Apache 2.0 license.
A Validator and Converter for the Synthetic Biology Open Language
This paper presents a new validation and conversion utility for the Synthetic Biology Open Language (SBOL). This utility can be accessed directly in software using the libSBOLj library, through a web interface, or using a web service via RESTful API calls. The validator checks all required and best practice rules set forth in the SBOL specification document, and it reports back to the user the location within the document of any errors found. The converter is capable of translating from/to SBOL 1, GenBank, and FASTA formats to/from SBOL 2. The SBOL Validator/Converter utility is released freely and open source under the Apache 2.0 license. The online version of the validator/converter utility can be found here: http://www.async.ece.utah.edu/sbol-validator/. The source code for the validator/converter can be found here: http://github.com/SynBioDex/SBOL-Validator/.
Model Discovery for Analog/Mixed-Signal Circuits
Advantages of formal verification are offset by the difficulties in the generating of good system models. This paper presents an improvement to the existing method, which generates models from a set of simulation traces. The proposed methodology aims to improve model precision by introducing new derivative based discretization method. Furthermore, a fine control over model fitness is provided via a notion of data rule mining, a novel approach to finding recurring patterns in the input data.
Invited: Advances in formal methods for the design of analog/mixed-signal systems
Analog/mixed-signal (AMS) systems are rapidly expanding in all domains of information and communication technology. They are a critical part of the support for large-scale high-performance digital systems, provide important functionalities in medium-scale embedded and mobile systems, and act as a core organ of autonomous electronics such as sensor nodes. Analog and digital parts are closely inter-mixed, hence demanding AMS design methods and tools to be more holistic. In particular, the emergence of “little digital” electronics inside or near analog circuitry calls for the increasing use of asynchronous logic. To cope with the growing complexity of AMS designs, formal methods are required to complement traditional simulation approaches. This paper presents an overview of the state-of-the-art in AMS formal verification and asynchronous design that enables the development of analog/asynchronous co-design methods. One such co-design methodology is exemplified by the LEMA-Workcraft workflow currently under development by the authors.
SBOLme: a Repository of SBOL Parts for Metabolic Engineering
The Synthetic Biology Open Language (SBOL) is a community-driven open language to promote standardization in synthetic biology. To support the use of SBOL in metabolic engineering, we developed SBOLme, the first open-access repository of SBOL 2-compliant biochemical parts for a wide range of metabolic engineering applications. The URL of our repository is http://www.cbrc.kaust.edu.sa/sbolme.
A standard-enabled workflow for synthetic biology
A synthetic biology workflow is composed of data repositories that provide information about genetic parts, sequence-level design tools to compose these parts into circuits, visualization tools to depict these designs, genetic design tools to select parts to create systems, and modeling and simulation tools to evaluate alternative design choices. Data standards enable the ready exchange of information within such a workflow, allowing repositories and tools to be connected from a diversity of sources. The present paper describes one such workflow that utilizes, among others, the Synthetic Biology Open Language (SBOL) to describe genetic designs, the Systems Biology Markup Language to model these designs, and SBOL Visual to visualize these designs. We describe how a standard-enabled workflow can be used to produce types of design information, including multiple repositories and software tools exchanging information using a variety of data standards. Recently, the ACS Synthetic Biology journal has recommended the use of SBOL in their publications.
Synthetic Biology Open Language (SBOL) Version 2.1.0
Synthetic biology builds upon the techniques and successes of genetics, molecular biology, and metabolic engineering by applying engineering principles to the design of biological systems. The field still faces substantial challenges, including long development times, high rates of failure, and poor reproducibility. One method to ameliorate these problems would be to improve the exchange of information about designed systems between laboratories. The Synthetic Biology Open Language (SBOL) has been developed as a standard to support the specification and exchange of biological design information in synthetic biology, filling a need not satisfied by other pre-existing standards. This document details version 2.1 of SBOL that builds upon version 2.0 published in last year’s JIB special issue. In particular, SBOL 2.1 includes improved rules for what constitutes a valid SBOL document, new role fields to simplify the expression of sequence features and how components are used in context, and new best practices descriptions to improve the exchange of basic sequence topology information and the description of genetic design provenance, as well as miscellaneous other minor improvements.
Specifications of Standards in Systems and Synthetic Biology: Status and Developments in 2016
Standards are essential to the advancement of science and technology. In systems and synthetic biology, numerous standards and associated tools have been developed over the last 16 years. This special issue of the Journal of Integrative Bioinformatics aims to support the exchange, distribution and archiving of these standards, as well as to provide centralised and easily citable access to them.
Toward Community Standards and Software for Whole-Cell Modeling
Objective: Whole-cell (WC) modeling is a promising tool for biological research, bioengineering, and medicine. However, substantial work remains to create accurate comprehensive models of complex cells. Methods: We organized the 2015 Whole-Cell Modeling Summer School to teach WC modeling and evaluate the need for new WC modeling standards and software by recoding a recently published WC model in the Systems Biology Markup Language. Results: Our analysis revealed several challenges to representing WC models using the current standards. Conclusion: We, therefore, propose several new WC modeling standards, software, and databases. Significance: We anticipate that these new standards and software will enable more comprehensive models.
Design of Mixed-Signal Systems With Asynchronous Control
This paper presents a novel workflow for the design of mixed-signal systems with asynchronous control. Current methods rely on synchronous control logic and full-system simulation, which might lead to suboptimal results and even project respins due to critical errors. Asynchronous circuits can provide greater robustness, reactivity, and power efficiency. The proposed workflow aims to combine state-of-the-art tools for asynchronous circuit design and formal verification of analog systems in a unified environment. The effectiveness of this methodology is demonstrated by the analysis of a buck converter.
Efficient Analysis of Systems Biology Markup Language Models of Cellular Populations Using Arrays
The Systems Biology Markup Language (SBML) has been widely used for modeling biological systems. Although SBML has been successful in representing a wide variety of biochemical models, the core standard lacks the structure for representing large complex regular systems in a standard way, such as whole-cell and cellular population models. These models require a large number of variables to represent certain aspects of these types of models, such as the chromosome in the whole-cell model and the many identical cell models in a cellular population. While SBML core is not designed to handle these types of models efficiently, the proposed SBML arrays package can represent such regular structures more easily. However, in order to take full advantage of the package, analysis needs to be aware of the arrays structure. When expanding the array constructs within a model, some of the advantages of using arrays are lost. This paper describes a more efficient way to simulate arrayed models. To illustrate the proposed method, this paper uses a population of repressilator and genetic toggle switch circuits as examples. Results show that there are memory benefits using this approach with a modest cost in runtime.
Sharing Structure and Function in Biological Design with SBOL 2.0
The Synthetic Biology Open Language (SBOL) is a standard that enables collaborative engineering of biological systems across different institutions and tools. SBOL is developed through careful consideration of recent synthetic biology trends, real use cases, and consensus among leading researchers in the field and members of commercial biotechnology enterprises. We demonstrate and discuss how a set of SBOL-enabled software tools can form an integrated, cross-organizational workflow to recapitulate the design of one of the largest published genetic circuits to date, a 4-input AND sensor. This design encompasses the structural components of the system, such as its DNA, RNA, small molecules, and proteins, as well as the interactions between these components that determine the system’s behavior/function. The demonstrated workflow and resulting circuit design illustrate the utility of SBOL 2.0 in automating the exchange of structural and functional specifications for genetic parts, devices, and the biological systems in which they operate.
A Converter from the Systems Biology Markup Language to the Synthetic Biology Open Language
Standards are important to synthetic biology because they enable exchange and reproducibility of genetic designs. This paper describes a procedure for converting between two standards: the Systems Biology Markup Language (SBML) and the Synthetic Biology Open Language (SBOL). SBML is a standard for behavioral models of biological systems at the molecular level. SBOL describes structural and basic qualitative behavioral aspects of a biological design. Converting SBML to SBOL enables a consistent connection between behavioral and structural information for a biological design. The conversion process described in this paper leverages Systems Biology Ontology (SBO) annotations to enable inference of a designs qualitative function.
Verification methodologies for fault-tolerant network-on-chip systems
Over the last decade, cyber-physical systems (CPSs) have seen significant applications in many safety-critical areas, such as autonomous automotive systems, automatic pilot avionics, wireless sensor networks, etc. A Cps uses networked embedded computers to monitor and control physical processes. The motivating example for this dissertation is the use of fault- tolerant routing protocol for a Network-on-Chip (NoC) architecture that connects electronic control units (Ecus) to regulate sensors and actuators in a vehicle. With a network allowing Ecus to communicate with each other, it is possible for them to share processing power to improve performance. In addition, networked Ecus enable flexible mapping to physical processes (e.g., sensors, actuators), which increases resilience to Ecu failures by reassigning physical processes to spare Ecus. For the on-chip routing protocol, the ability to tolerate network faults is important for hardware reconfiguration to maintain the normal operation of a system. Adding a fault-tolerance feature in a routing protocol, however, increases its design complexity, making it prone to many functional problems. Formal verification techniques are therefore needed to verify its correctness. This dissertation proposes a link-fault-tolerant, multiflit wormhole routing algorithm, and its formal modeling and verification using two different methodologies. An improvement upon the previously published fault-tolerant routing algorithm, a link-fault routing algorithm is proposed to relax the unrealistic node-fault assumptions of these algorithms, while avoiding deadlock conservatively by appropriately dropping network packets. This routing algorithm, together with its routing architecture, is then modeled in a process-algebra language LNT, and compositional verification techniques are used to verify its key functional properties. As a comparison, it is modeled using channel-level VHDL which is compiled to labeled Petri-nets (LPNs). Algorithms for a partial order reduction method on LPNs are given. An optimal result is obtained from heuristics that trace back on LPNs to find causally related enabled predecessor transitions. Key observations are made from the comparison between these two verification methodologies.
Engineering Genetic Circuits
An Introduction to Systems BioengineeringTakes a Clear and Systematic Engineering Approach to Systems BiologyFocusing on genetic regulatory networks, Engineering Genetic Circuits presents the modeling, analysis, and design methods for systems biology. It discusses how to examine experimental data to learn about mathematical models, develop efficien
An improved fault-tolerant routing algorithm for a Network-on-Chip derived with formal analysis
A fault-tolerant routing algorithm in Network-on-Chip (NoC) architectures provides adaptivity for on-chip communications. Adding fault-tolerance adaptivity to a routing algorithm increases its design complexity and makes it prone to deadlock and other problems if improperly implemented. Formal verification techniques are needed to check the correctness of the design. This paper describes the discovery of a potential livelock problem through formal analysis on an extension of the link-fault tolerant NoC architecture introduced by Wu et al. In the process of eliminating this problem, an improved routing architecture is derived. The improvement simplifies the routing architecture, enabling successful verification using the CADP verification toolbox. The routing algorithm is proven to have several desirable properties including deadlock and livelock freedom, and tolerance to a single-link-fault.
Stochastic Model Checking of Genetic Circuits
Synthetic genetic circuits have a number of exciting potential applications such as cleaning up toxic waste, hunting and killing tumor cells, and producing drugs and bio-fuels more efficiently. When designing and analyzing genetic circuits, researchers are often interested in the probability of observing certain behaviors. Discerning these probabilities typically involves simulating the circuit to produce some time series data and computing statistics over the resulting data. However, for very rare behaviors of complex genetic circuits, it becomes computationally intractable to obtain good results as the number of required simulation runs grows exponentially. It is, therefore, necessary to apply numerical methods to determine these probabilities directly. This article describes how stochastic model checking, a method for determining the likelihood that certain events occur in a system, can by applied to models of genetic circuits by translating them into continuous-time Markov chains (CTMCs) and analyzing them using Markov chain analysis to check continuous stochastic logic (CSL) properties. The utility of this approach is demonstrated with several case studies illustrating how this method can be used to perform design space exploration of two genetic oscillators and two genetic state-holding elements. Our results show that this method results in a substantial speedup as compared with conventional simulation-based approaches.
Introduction to the Special Issue on Computational Synthetic Biology
The goal of this special issue is to introduce the field of computational synthetic biology to engineers and computer scientists. The first article gives an introduction to the key biological principles and experimental techniques that support synthetic biology, and it draws analogies with the computing field. This issue also includes five original research articles in computational synthetic biology. The first research article discusses how standards can be used to modularize the design process for genetic circuits. The next two articles introduce new abstraction techniques to improve the efficiency of analysis of genetic circuit models. The last two articles introduce new design techniques that help decouple design from construction. We hope this sampling from the field will help to motivate others to join this exciting and rich area of research.
SBOL Visual: A Graphical Language for Genetic Designs
Synthetic Biology Open Language (SBOL) Visual is a graphical standard for genetic engineering. It consists of symbols representing DNA subsequences, including regulatory elements and DNA assembly features. These symbols can be used to draw illustrations for communication and instruction, and as image assets for computer-aided design. SBOL Visual is a community standard, freely available for personal, academic, and commercial use (Creative Commons CC0 license). We provide prototypical symbol images that have been used in scientific publications and software tools. We encourage users to use and modify them freely, and to join the SBOL Visual community: http://www.sbolstandard.org/visual.
libSBOLj 2.0: A Java Library to Support SBOL 2.0
The Synthetic Biology Open Language (SBOL) is an emerging data standard for representing synthetic biology designs. The goal of SBOL is to improve the reproducibility of these designs and their electronic exchange between researchers and/or genetic design automation tools. The latest version of the standard, SBOL 2.0, enables the annotation of a large variety of biological components (e.g., DNA, RNA, proteins, complexes, small molecules, etc.) and their interactions. SBOL 2.0 also allows researchers to organize components into hierarchical modules, to specify their intended functions, and to link modules to models that describe their behavior mathematically. To support the use of SBOL 2.0, we have developed the libSBOLj 2.0 Java library, which provides an easy to use Application Programming Interface (API) for developers, including manipulation of SBOL constructs, serialization to and from an RDF/XML file format, and migration support in the form of conversion from the prior SBOL 1.1 standard to SBOL 2.0. This letter describes the libSBOLj 2.0 library and key engineering decisions involved in its design.
Systems Biology Markup Language (SBML) Level 2 Version 5: Structures and Facilities for Model Definitions
Computational models can help researchers to interpret data, understand biological function, and make quantitative predictions. The Systems Biology Markup Language (SBML) is a file format for representing computational models in a declarative form that can be exchanged between different software systems. SBML is oriented towards describing biological processes of the sort common in research on a number of topics, including metabolic pathways, cell signaling pathways, and many others. By supporting SBML as an input/output format, different tools can all operate on an identical representation of a model, removing opportunities for translation errors and assuring a common starting point for analyses and simulations. This document provides the specification for Version 5 of SBML Level 2. The specification defines the data structures prescribed by SBML as well as their encoding in XML, the eXtensible Markup Language. This specification also defines validation rules that determine the validity of an SBML document, and provides many examples of models in SBML form. Other materials and software are available from the SBML project web site, http://sbml.org/.
Synthetic Biology Open Language (SBOL) Version 2.0.0
Synthetic biology builds upon the techniques and successes of genetics, molecular biology, and metabolic engineering by applying engineering principles to the design of biological systems. The field still faces substantial challenges, including long development times, high rates of failure, and poor reproducibility. One method to ameliorate these problems would be to improve the exchange of information about designed systems between laboratories. The Synthetic Biology Open Language (SBOL) has been developed as a standard to support the specification and exchange of biological design information in synthetic biology, filling a need not satisfied by other pre-existing standards. This document details version 2.0 of SBOL, introducing a standardized format for the electronic exchange of information on the structural and functional aspects of biological designs. The standard has been designed to support the explicit and unambiguous description of biological designs by means of a well defined data model. The standard also includes rules and best practices on how to use this data model and populate it with relevant design details. The publication of this specification is intended to make these capabilities more widely accessible to potential developers and users in the synthetic biology community and beyond.
Generating Systems Biology Markup Language Models from the Synthetic Biology Open Language
In the context of synthetic biology, model generation is the automated process of constructing biochemical models based on genetic designs. This paper discusses the use cases for model generation in genetic design automation (GDA) software tools and introduces the foundational concepts of standards and model annotation that make this process useful. Finally, this paper presents an implementation of model generation in the GDA software tool iBioSim and provides an example of generating a Systems Biology Markup Language (SBML) model from a design of a 4-input AND sensor written in the Synthetic Biology Open Language (SBOL).
Efficient, Sound Formal Verification for Analog/Mixed-Signal Circuits
The increasing demand for smaller, more efficient circuits has created a need for both digital and analog designs to scale down. Digital technologies have been successful in meeting this challenge, but analog circuits have lagged behind due to smaller transistor sizes having a disproportionate negative affect. Since many applications require small, low-power analog circuits, the trend has been to take advantage of digital’s ability to scale by replacing as much of the analog circuitry as possible with digital counterparts. The results are known as emphdigitally-intensive analog/mixed-signal (AMS) circuits. Though such circuits have helped the scaling problem, they have further complicated verification. This dissertation improves on techniques for AMS property specifications, as well as, develops sound, efficient extensions to formal AMS verification methods. With the emphlanguage for analog/mixed-signal properties (LAMP), one has a simple intuitive language for specifying AMS properties. LAMP provides a more procedural method for describing properties that is more straightforward than temporal logic-like languages. However, LAMP is still a nascent language and is limited in the types of properties it is capable of describing. This dissertation extends LAMP by adding statements to ignore transient periods and be able to reset the property check when the environment conditions change. After specifying a property, one needs to verify that the circuit satisfies the property. An efficient method for formally verifying AMS circuits is to use the restricted polyhedral class of emphzones. Zones have simple operations for exploring the reachable state space, but they are only applicable to circuit models that utilize constant rates. To extend zones to more general models, this dissertation provides the theory and implementation needed to soundly handle models with ranges of rates. As a second improvement to the state representation, this dissertation describes how octagons can be adapted to model checking AMS circuit models. Though zones have efficient algorithms, it comes at a cost of over-approximating the reachable state space. Octagons have similarly efficient algorithms while adding additional flexibility to reduce the necessary over-approximations. Finally, the full methodology described in this dissertation is demonstrated on two examples. The first example is a switched capacitor integrator that has been studied in the context of transforming the original formal model to use only single rate assignments. Th property of not saturating is written in LAMP, the circuit is learned, and the property is checked against a faulty and correct circuit. In addition, it is shown that the zone extension, and its implementation with octagons, recovers all previous conclusions with the switched capacitor integrator without the need to translate the model. In particular, the method applies generally to all the models produced and does not require the soundness check needed by the translational approach to accept positive verification results. As a second example, the full tool flow is demonstrated on a digital C-element that is driven by a pair of RC networks, creating an AMS circuit. The RC networks are chosen so that the inputs to the C-element are ordered. LAMP is used to codify this behavior and it is verified that the input signals change in the correct order for the provided SPICE simulation traces.
Specifications of Standards in Systems and Synthetic Biology
Standards shape our everyday life. From nuts and bolts to electronic devices and technological processes, standardised products and processes are all around us. Standards have technological and economic benefits, such as making information exchange, production, and services more efficient. However, novel, innovative areas often either lack proper standards, or documents about standards in these areas are not available from a centralised platform or formal body (such as the International Standardisation Organisation).textless/ptextgreatertextlessptextgreaterSystems and synthetic biology is a relatively novel area, and it is only in the last decade that the standardisation of data, information, and models related to systems and synthetic biology has become a community-wide effort. Several open standards have been established and are under continuous development as a community initiative. COMBINE, the ‘COmputational Modeling in BIology’ NEtwork [1] has been established as an umbrella initiative to coordinate and promote the development of the various community standards and formats for computational models. There are yearly two meeting, HARMONY (Hackathons on Resources for Modeling in Biology), Hackathon-type meetings with a focus on development of the support for standards, and COMBINE forums, workshop-style events with oral presentations, discussion, poster, and breakout sessions for further developing the standards. For more information see http://co.mbine.org/. So far the different standards were published and made accessible through the standards’ web-pages or preprint services. The aim of this special issue is to provide a single, easily accessible and citable platform for the publication of standards in systems and synthetic biology. This special issue is intended to serve as a central access point to standards and related initiatives in systems and synthetic biology, it will be published annually to provide an opportunity for standard development groups to communicate updated specifications.
SBML Level 3 package: Hierarchical Model Composition, Version 1 Release 3
Constructing a model in a hierarchical fashion is a natural approach to managing model complexity, and offers additional opportunities such as the potential to re-use model components. The SBML Level 3 Version 1 Core specification does not directly provide a mechanism for defining hierarchical models, but it does provide a mechanism for SBML packages to extend the Core specification and add additional syntactical constructs. The SBML Hierarchical Model Composition package for SBML Level 3 adds the necessary features to SBML to support hierarchical modeling. The package enables a modeler to include submodels within an enclosing SBML model, delete unneeded or redundant elements of that submodel, replace elements of that submodel with element of the containing model, and replace elements of the containing model with elements of the submodel. In addition, the package defines an optional “port” construct, allowing a model to be defined with suggested interfaces between hierarchical components; modelers can chose to use these interfaces, but they are not required to do so and can still interact directly with model elements if they so chose. Finally, the SBML Hierarchical Model Composition package is defined in such a way that a hierarchical model can be “flattened” to an equivalent, non-hierarchical version that uses only plain SBML constructs, thus enabling software tools that do not yet support hierarchy to nevertheless work with SBML hierarchical models.
Compositional Model Checking of Concurrent Systems
This paper presents a compositional framework to address the state explosion problem in model checking of concurrent systems. This framework takes as input a system model described as a network of communicating components in a high-level description language, finds the local state transition models for each individual component where local properties can be verified, and then iteratively reduces and composes the component state transition models to form a reduced global model for the entire system where global safety properties can be verified. The state space reductions used in this framework result in a reduced model that contains the exact same set of observably equivalent executions as in the original model, therefore, no false counter-examples result from the verification of the reduced model. This approach allows designs that cannot be handled monolithically or with partial-order reduction to be verified without difficulty. The experimental results show significant scale-up of this compositional verification framework on a number of non-trivial concurrent system models.
Reachability Analysis Using Extremal Rates
General hybrid systems can be difficult to verify due to their generality. To reduce the complexity, one often specializes to hybrid systems where the complexity is more manageable. If one reduces the modeling formalism to ones where the continuous variables have a single rate, then it may be possible to use the methods of zones to find the reachable state space. Zones are a restricted class of polyhedra formed by considering the intersections of half-planes defined by two variable constraints. Due to their simplicity, zones have simpler, more efficient methods of manipulation than more general polyhedral classes, though they are less accurate. This paper extends the method of zones to labeled Petri net (LPN) models with continuous variables that evolve over a range of rates.
The Synthetic Biology Open Language
This volume provides complete coverage of the computational approaches currently used in Synthetic Biology. Chapters focus on computational methods and algorithms for the design of bio-components, insight on CAD programs, analysis techniques, and distributed systems. Written in the highly successful Methods in Molecular Biology series format, the chapters include the kind of detailed description and implementation advice that is crucial for getting optimal results in the laboratory. Authoritative and practical, Computational Methods in Synthetic Biology serves as a guide to plan in silico the in vivo or in vitro construction of a variety of synthetic bio-circuits.
Promoting Coordinated Development of Community-Based Information Standards for Modeling in Biology: The COMBINE Initiative
COMBINE (Computational Modeling in Biology Network) is a consortium of groups involved in the development of open community standards and formats used in computational modeling in biology. COMBINE’s aim is to act as a coordinator, facilitator, and resource for different standardization efforts whose domains of use cover related areas of the computational biology space. In this perspective article, we summarize COMBINE, its general organization, and the community standards and other efforts involved in it. Our goals are to help guide readers toward standards that may be suitable for their research activities, as well as to direct interested readers to relevant communities where they can best expect to receive assistance in how to develop interoperable computational models.
Efficient Analysis Methods in Synthetic Biology
This volume provides complete coverage of the computational approaches currently used in Synthetic Biology. Chapters focus on computational methods and algorithms for the design of bio-components, insight on CAD programs, analysis techniques, and distributed systems. Written in the highly successful Methods in Molecular Biology series format, the chapters include the kind of detailed description and implementation advice that is crucial for getting optimal results in the laboratory. Authoritative and practical, Computational Methods in Synthetic Biology serves as a guide to plan in silico the in vivo or in vitro construction of a variety of synthetic bio-circuits.
Computational Synthetic Biology: Progress and the Road Ahead
Synthetic biology promises to leverage engineering principles to enable model-based design of genetic circuits. To be successful, advancements are needed in both experimental and computational methods to support this new approach. This paper focuses on the progress on the computational side, the remaining challenges, and the road ahead. While much work remains to be done for computational methods to truly have impact on experimental synthetic biology, collaborations between computational and experimental synthetic biologists have the potential for tremendous impact in the areas of health, energy, and the environment.
A new assertion property language for analog/mixed-signal circuits
This book brings together a selection of the best papers from the sixteenth edition of the Forum on specification and Design Languages Conference (FDL), which was held in September 2013 in Paris, France. FDL is a well-established international forum devoted to dissemination of research results, practical experiences and new ideas in the application of specification, design and verification languages to the design, modeling and verification of integrated circuits, complex hardware/software embedded systems and mixed-technology systems.
Technology mapping of genetic circuit designs
Synthetic biology is a new field in which engineers, biologists, and chemists are working together to transform genetic engineering into an advanced engineering discipline, one in which the design and construction of novel genetic circuits are made possible through the application of engineering principles. This dissertation explores two engineering strategies to address the challenges of working with genetic technology, namely the development of standards for describing genetic components and circuits at separate yet connected levels of detail and the use of Genetic Design Automation (GDA) software tools to simplify and speed up the process of optimally designing genetic circuits. Its contributions to the field of synthetic biology include (1) a proposal for the next version of the Synthetic Biology Open Language (SBOL), an existing standard for specifying and exchanging genetic designs electronically, and (2) a GDA work ow that enables users of the software tool iBioSim to create an abstract functional specication, automatically select genetic components that satisfy the specication from a design library, and compose the selected components into a standardized genetic circuit design for subsequent analysis and physical construction. Ultimately, this dissertation demonstrates how existing techniques and concepts from electrical and computer engineering can be adapted to overcome the challenges of genetic design and is an example of what is possible when working with publicly available standards for genetic design.
Formal Analysis of a Fault-Tolerant Routing Algorithm for a Network-on-Chip
A fault-tolerant routing algorithm in Network-on-Chip architectures provides adaptivity for on-chip communications. Adding fault-tolerance adaptivity to a routing algorithm increases its design complexity and makes it prone to deadlock and other problems if improperly implemented. Formal verification techniques are needed to check the correctness of the design. This paper performs formal analysis on an extension of the link-fault tolerant Network-on-Chip architecture introduced by Wu et al that supports multiflit wormhole routing. This paper describes several lessons learned during the process of constructing a formal model of this routing architecture. Finally, this paper presents how the deadlock freedom and tolerance to a single-link fault is verified for a two-by-two mesh version of this routing architecture.
Directed Acyclic Graph-Based Technology Mapping of Genetic Circuit Models
As engineering foundations such as standards and abstraction begin to mature within synthetic biology, it is vital that genetic design automation (GDA) tools be developed to enable synthetic biologists to automatically select standardized DNA components from a library to meet the behavioral specification for a genetic circuit. To this end, we have developed a genetic technology mapping algorithm that builds on the directed acyclic graph (DAG) based mapping techniques originally used to select parts for digital electronic circuit designs and implemented it in our GDA tool, iBioSim. It is among the first genetic technology mapping algorithms to adapt techniques from electronic circuit design, in particular the use of a cost function to guide the search for an optimal solution, and perhaps that which makes the greatest use of standards for describing genetic function and structure to represent design specifications and component libraries. This paper demonstrates the use of our algorithm to map the specifications for three different genetic circuits against four randomly generated libraries of increasing size to evaluate its performance against both exhaustive search and greedy variants for finding optimal and near-optimal solutions.
LEMA: A tool for the formal verification of digitally-intensive analog/mixed-signal circuits
The increasing integration of analog/mixed-signal (AMS) circuits into system designs has further complicated an already difficult verification problem. Recently, formal verification, which has been successful in the purely digital domain, has made some in-roads in the AMS domain. This paper describes one such formal verification tool for AMS circuits, LEMA. In particular, LEMA is capable of generating a formal model from simulation traces that, when coupled with a formal property provided in our new property language, can be model checked with one of three model checkers within LEMA. This paper briefly describes the capabilities of the LEMA AMS verification tool flow.
Hierarchical Stochastic Simulation of Genetic Circuits
This thesis describes a hierarchical stochastic simulation algorithm which has been implemented within iBioSim, a tool used to model, analyze, and visualize genetic circuits. Many biological analysis tools flatten out hierarchy before simulation, but there are many disadvantages associated with this approach. First, the memory required to represent the model can quickly expand in the process. Second, the flattening process is computationally expensive. Finally, when modeling a dynamic cellular population within iBioSim, inlining the hierarchy of the model is inefficient since models must grow dynamically over time. This paper discusses a new approach to handle hierarchy on the fly to make the tool faster and more memory-efficient. This approach yields significant performance improvements as compared to the former flat analysis method.
Hierarchical stochastic simulation of genetic circuits
This paper describes a hierarchical stochastic simulation algorithm which has been implemented within iBioSim, a tool used to model, analyze, and visualize genetic circuits. Many biological analysis tools flatten out hierarchy before simulation, but there are many disadvantages associated with this approach. First, the memory required to represent the model can quickly expand in the process. Second, the flattening process is computationally expensive. Finally, when modeling a dynamic cellular population within iBioSim, inlining the hierarchy of the model is inefficient since models must grow dynamically over time. This paper discusses a new approach to handle hierarchy on the fly to make the tool faster and more memory-efficient. This approach yields significant performance improvements as compared to the former flat analysis method.
Meeting report from the fourth meeting of the Computational Modeling in Biology Network (COMBINE)
The Computational Modeling in Biology Network (COMBINE) is an initiative to coordinate the development of community standards and formats in computational systems biology and related fields. This report summarizes the topics and activities of the fourth edition of the annual COMBINE meeting, held in Paris during September 16-20 2013, and attended by a total of 96 people. This edition pioneered a first day devoted to modeling approaches in biology, which attracted a broad audience of scientists thanks to a panel of renowned speakers. During subsequent days, discussions were held on many subjects including the introduction of new features in the various COMBINE standards, new software tools that use the standards, and outreach efforts. Significant emphasis went into work on extensions of the SBML format, and also into community-building. This year’s edition once again demonstrated that the COMBINE community is thriving, and still manages to help coordinate activities between different standards in computational systems biology.
A Methodology to Annotate Systems Biology Markup Language Models with the Synthetic Biology Open Language
Recently, we have begun to witness the potential of synthetic biology, noted here in the form of bacteria and yeast that have been genetically engineered to produce biofuels, manufacture drug precursors, and even invade tumor cells. The success of these projects, however, has often failed in translation and application to new projects, a problem exacerbated by a lack of engineering standards that combine descriptions of the structure and function of DNA. To address this need, this paper describes a methodology to connect the systems biology markup language (SBML) to the synthetic biology open language (SBOL), existing standards that describe biochemical models and DNA components, respectively. Our methodology involves first annotating SBML model elements such as species and reactions with SBOL DNA components. A graph is then constructed from the model, with vertices corresponding to elements within the model and edges corresponding to the cause-and-effect relationships between these elements. Lastly, the graph is traversed to assemble the annotating DNA components into a composite DNA component, which is used to annotate the model itself and can be referenced by other composite models and DNA components. In this way, our methodology can be used to build up a hierarchical library of models annotated with DNA components. Such a library is a useful input to any future genetic technology mapping algorithm that would automate the process of composing DNA components to satisfy a behavioral specification. Our methodology for SBML-to-SBOL annotation is implemented in the latest version of our genetic design automation (GDA) software tool, iBioSim.
Hierarchical Stochastic Simulation Algorithm for SBML Models of Genetic Circuits
This paper describes a hierarchical stochastic simulation algorithm which has been implemented within iBioSim, a tool used to model, analyze, and visualize genetic circuits. Many biological analysis tools flatten out hierarchy before simulation, but there are many disadvantages associated with this approach. First, the memory required to represent the model can quickly expand in the process. Second, the flattening process is computationally expensive. Finally, when modeling a dynamic cellular population within iBioSim, inlining the hierarchy of the model is inefficient since models must grow dynamically over time. This paper discusses a new approach to handle hierarchy on the fly to make the tool faster and more memory-efficient. This approach yields significant performance improvements as compared to the former flat analysis method.
Platforms for Genetic Design Automation
Crucial to the success of synthetic biology is the development of platforms for genetic design automation (GDA). This chapter presents the current state-of-the-art in GDA tools. This chapter also briefly describes the standards used for data representation that enable these GDA tools to work together to complete a genetic design task and the emerging repositories that are available to archive and share these data. Finally, this chapter compares tool capabilities and discusses future requirements for a complete GDA workflow.
A new assertion property language for analog/mixed-signal circuits
In automating the verification of analog/mixed-signal (AMS) circuits, it essential to have a specification language that can describe the behavior that needs to be checked. Although powerful and very expressive, many such languages have a steep learning curve for designers and are complicated to use. This paper describes a simpler, more intuitive language called the Language for Analog/Mixed-Signal Properties (LAMP) that is incorporated into our LEMA verification tool, and demonstrates how this language can be used for AMS verification.
Stochastic Analysis of Synthetic Genetic Circuits
Over the past few decades, synthetic biology has generated great interest to biologists and engineers alike. Synthetic biology combines the research of biology with the engineering principles of standards, abstraction, and automated construction with the ultimate goal of being able to design and build useful biological systems. To realize this goal, researchers are actively working on better ways to model and analyze synthetic genetic circuits, groupings of genes that influence the expression of each other through the use of proteins. When designing and analyzing genetic circuits, researchers are often interested in building circuits that exhibit a particular behavior. Usually, this involves simulating their models to produce some time series data and analyzing this data to discern whether or not the circuit behaves appropriately. This method becomes less attractive as circuits grow in complexity because it becomes very time consuming to generate a sufficient amount of runs for analysis. In addition, trying to select representative runs out of a large data set is tedious and error-prone thereby motivating methods of automating this analysis. This has led to the need for design space exploration techniques that allow synthetic biologists to easily explore the effect of varying parameters and efficiently consider alternative designs of their systems. This dissertation attempts to address this need by proposing new analysis and verification techniques for synthetic genetic circuits. In particular, it applies formal methods such as model checking techniques to models of genetic circuits in order to ensure that they behave correctly and are as robust as possible for a variety of different inputs and/or parameter settings. However, model checking stochastic systems is not as simple as model checking deterministic systems where it is always known what the next state of the system will be at any given step. Stochastic systems can exhibit a variety of different behaviors that are chosen randomly with different probabilities at each time step. Therefore, model checking a stochastic system involves calculating the probability that the system will exhibit a desired behavior. Although it is often more difficult to work with the probabilities that stochastic systems introduce, stochastic systems and the models that represent them are becoming commonplace in many disciplines including electronic circuit design where as parts are being made smaller and smaller, they are becoming less reliable. In addition to stochastic model checking, this dissertation proposes a new incremental stochastic simulation algorithm (iSSA) based on Gillespie’s stochastic simulation algorithm (SSA) that is capable of presenting a researcher with a simulation trace of the typical behavior of the system. Before the development of this algorithm, discerning this information was extremely error-prone as it involved performing many simulations and attempting to wade through the massive amounts of data. This algorithm greatly aids researchers in designing genetic circuits as it efficiently shows the researcher the most likely behavior of the circuit. Both the iSSA and stochastic model checking can be used in concert to give a researcher the likelihood that the system will exhibit its most typical behavior. Once the typical behavior is known, properties for nontypical behaviors can be constructed and their likelihoods can also be computed. This methodology is applied to several genetic circuits leading to new understanding of the effects of various parameters on the behavior of these circuits.
Improved Model Generation and Property Specification for Analog/Mixed-Signal Circuits
This document describes an improved method of formal verification of complex analog/mixed-signal (AMS) circuits. Currently, in our LEMA tool, verification properties are encoded using labeled Petri net (LPN). These LPNs are generated manually, a tedious process that requires the user to have considerable familiarity with the tool. To eliminate this time-consuming process, our LEMA tool is extended to include a translator that converts properties written in a property specification language to LPNs. New methods are also implemented to separate the transient period from the stable output period, thus improving the generated model. Also, the current methodology generates the circuit models for the input values used during the simulation of the circuit. So, models generated for other control input values are not accurate. In this case, accuracy of the generated models is improved by using a linear abstraction method like interpolation.
Verification of digitally-intensive analog circuits via kernel ridge regression and hybrid reachability analysis
The emergence of digitally-intensive analog circuits introduces new challenges to formal verification due to increased digital design content, and non-ideal digital effects such as finite resolution, round-off error and overflow. We propose a machine learning approach to convert digital blocks to conservative analog approximations via the use of kernel ridge regression. These learned models are then adopted in a hybrid formal reachability analysis framework where the support function based manipulations are developed to efficiently handle the large linear portion of the design and the more general satisfiability modulo theories technique is applied to the remaining nonlinear portion. The efficiency of the proposed method is demonstrated for the locked time verification of a digitally intensive phase locked loop.
Dynamic Modeling of Cellular Populations within iBioSim
As the complexity of synthetic genetic circuits increases, modeling is becoming a necessary first step to inform subsequent experimental efforts. In recent years, the design automation community has developed a wealth of computational tools for assisting experimentalists in designing and analyzing new genetic circuits at several scales. However, existing software primarily caters to either the DNA- or single-cell level, with little support for the multicellular level. To address this need, the iBioSim software package has been enhanced to provide support for modeling, simulating, and visualizing dynamic cellular populations in a two-dimensional space. This capacity is fully integrated into the software, capitalizing on iBioSim’s strengths in modeling, simulating, and analyzing single-celled systems.
Modeling and design automation of biological circuits and systems
Circuit designers are increasingly more drawn to challenges in modeling and designing biological circuits and systems. While the principles of biological organization and architecture resemble those in systems that engineers are designing, the complexity of biological systems still seems to be beyond the designed ones. This session discusses state-of-the-art in tackling such challenges, and presents existing methods for automation of model development, design and analysis of biological circuits and systems. The speakers are experts from systems biology, synthetic biology, and design automation fields. The three talks will cover a range of topics that include rule-based modeling approach to model cell signaling networks, automation of genetic circuit design, and the importance and development of standards in synthetic biology.
Using decision diagrams to compactly represent the state space for explicit model checking
The enormous number of states reachable during explicit model checking is the main bottleneck for scalability. This paper presents approaches of using decision diagrams to represent very large state space compactly and efficiently. This is possible for asynchronous systems as two system states connected by a transition often share many same local portions. Using decision diagrams can significantly reduce memory demand by not using memory to store the redundant information among different states. This paper considers multi-value decision diagrams for this purpose. Additionally, a technique to reduce the runtime overhead of using these diagrams is also described. Experimental results and comparison with the state compression method as implemented in the model checker SPIN show that the approaches presented in this paper are memory efficient for storing large state space with acceptable runtime overhead.
An Improvement in Partial Order Reduction Using Behavioral Analysis
Efficacy of partial order reduction in reducing state space relies on adequate extraction of the independence relation among possible behaviors. However, traditional approaches by statically analyzing system model structures are often not able to reveal enough independence for reduction. To address such a problem, this paper presents a behavioral analysis approach that uses a compositional reachability analysis method to generate the over-approximate local state spaces for all modules in a system where a much more precise independence relation can be extracted for partial order reduction. Compared to the static analysis approaches, significantly higher reduction on complexity can be seen in a number of non-trivial examples, and as a consequence, dramatically less time and memory are required to finish these examples.
A Compositional Minimization Approach for Large Asynchronous Design Verification
This paper presents a compositional minimization approach with efficient state space reductions for verifying non-trivial asynchronous designs. These reductions can result in a reduced model that contains the exact same set of observably equivalent behavior in the original model, therefore no false counter-examples result from the verification of the reduced model. This approach allows designs that cannot be handled monolithically or with partial-order reduction to be verified without difficulty. The experimental results show significant scale-up of the compositional minimization approach using these reductions on a number of large asynchronous designs.
Design and Test of Genetic Circuits Using tt iBioSim
The simulation of biological systems prior to their physical implementation can save time, money, and potentially provide insights into alternate designs. This paper presents a simulation environment which allows for a visual design process ultimately leading to a formal model which can be efficiently simulated.
Utilizing stochastic model checking to analyze genetic circuits
When designing and analyzing genetic circuits, researchers are often interested in the probability of the system reaching a given state within a certain amount of time. Usually, this involves simulating the system to produce some time series data and analyzing this data to discern the state probabilities. However, as the complexity of models of genetic circuits grow, it becomes more difficult for researchers to reason about the different states by looking only at time series simulation results of the models. To address this problem, this paper employs the use of stochastic model checking, a method for determining the likelihood that certain events occur in a system, with continuous stochastic logic (CSL) properties to obtain similar results. This goal is accomplished by the introduction of a methodology for converting a genetic circuit model (GCM) into a continuous-time Markov chain (CTMC). This CTMC is analyzed using transient Markov chain analysis to determine the likelihood that the circuit satisfies a given CSL property in a finite amount of time. This paper illustrates a use of this methodology to determine the likelihood of failure in a genetic toggle switch and compares these results to stochastic simulation-based analysis of this same circuit. Our results show that this method results in a substantial speedup as compared with conventional simulation-based approaches.
A Fault-Tolerant Routing Algorithm for a Network-on-Chip Using a Link Fault Model
Adaptive routing is a sensible approach to enhance fault-tolerance in Network-on-Chip (NoC) architectures, but can cause deadlock if implemented improperly. Glass and Ni proposed an adaptive routing algorithm based on a turn model which is proven to tolerate at least one fault in each routing process and is free of deadlock. However, when faults happen on links of a network instead of on nodes, these two claims are no longer true. This paper proposes an improved routing algorithm based on the Glass/Ni protocol which tolerates a single link fault while still avoiding deadlock in a mesh network. Simulation results indicate that this improved algorithm provides significant improvements in network reliability with minimal cost.
Erlang-delayed stochastic chemical kinetic formalism for efficient analysis of biological systems with non-elementary reaction effects
Stochastic chemical kinetics (SCK) has become an important formalism for modeling and analysis of complex biological systems as it can capture the discreteness and the randomness of underlying biochemical reactions. One of the assumptions made by SCK is that each reaction be an elementary step which cannot be broken down into smaller steps. As such, transition events of the SCK model occur spontaneously in that there is no time lag between the reaction initiation and completion. In practice, however, it is very difficult to experimentally determine if an observed state change is a result of an elementary reaction or a sequence of several reaction steps. To test various hypotheses on such time-delays through the use of the SCK, all of the intermediate reactions and species need to be explicitly specified, which can quickly become cumbersome. To more efficiently model and analyze potential effects of such intermediate reaction steps, this paper proposes a new formalism for higher-level discrete-stochastic treatment of biological systems with reaction delays. Our new formalism can represent a time delay caused by intermediate reaction steps as an Erlang random variable, allowing a model’s size to be reduced substantially. This paper illustrates an application of this formalism for analysis of the effect of different transcription elongation steps and rates on the distribution of RNA molecules.
Modeling and Visualization of Synthetic Genetic Circuits
The iBioSim tool is being developed to facilitate the construction and simulation of synthetic genetic circuits. In this project, we have created a user interface that is similar to those used to construct schematic diagrams which are familiar to electrical engineers. Promoters, chemical species, and biological relationships can be placed visually on a schematic diagram. Another enhancement was the creation of a new simulation visualization tool which allows the user to associate chemical species with color schemes, opacity, and cell size. This tool allows the user to see the cells behavior as if through a microscope.
Automated Abstraction of Labeled Petri Nets
Due to the increasing use and complexity of embedded and cyber-physical systems, proper validation of these systems is an increasingly important topic. Because these systems are used in safety-critical situations, even intermittent failures are unacceptable. Formal verification allows for comprehensive validation, but becomes unrealistically complicated for large models. This paper proposes certain transforms to simplify system models without removing any of the details required for verification. These details must be conservative, in that they may not remove any state from the model’s behavior. These transforms have shown this method promising in reducing the complexity of embedded system verification. I.
Verification of Analog/Mixed-Signal Circuits Using Labeled Hybrid Petri Nets
Mixed-signal designs integrate digital and analog circuits which complicates the already difficult verification problem. This paper presents a model, labeled hybrid Petri nets (LHPNs), that is developed to model this heterogeneous set of components. To support formal verification, this paper presents an efficient zone-based state space exploration algorithm for LHPNs. This algorithm uses a process known as warping which allows zones to describe continuous variables changing at variable rates. Finally, this paper describes the application of this algorithm to analog/mixed-signal circuit examples.
Learning Genetic Regulatory Network Connectivity from Time Series Data
Recent experimental advances facilitate the collection of time series data that indicate which genes in a cell are expressed. This information can be used to understand the genetic regulatory network that generates the data. Typically, Bayesian analysis approaches are applied which neglect the time series nature of the experimental data, have difficulty in determining the direction of causality, and do not perform well on networks with tight feedback. To address these problems, this paper presents a method to learn genetic network connectivity which exploits the time series nature of experimental data to achieve better causal predictions. This method first breaks up the data into bins. Next, it determines an initial set of potential influence vectors for each gene based upon the probability of the gene’s expression increasing in the next time step. These vectors are then combined to form new vectors with better scores. Finally, these influence vectors are competed against each other to determine the final influence vector for each gene. The result is a directed graph representation of the genetic network’s repression and activation connections. Results are reported for several synthetic networks with tight feedback showing significant improvements in recall and runtime over Yu’s dynamic Bayesian approach. Promising preliminary results are also reported for an analysis of experimental data for genes involved in the yeast cell cycle.
Automatic Extraction of Behavioral Models from Simulations of Analog/Mixed-Signal (AMS) Circuits
Verification of analog circuits is becoming a bottle-neck for the verification of complex analog/mixed-signal (AMS) circuits. In order to assist functional verification of complex AMS system-on-chips (SoCs), there is a need to represent the transistor-level circuits in the form of abstract models. The ability to represent the analog circuits as behavioral models is necessary, but not sufficient. Though there exist languages like Verilog-AMS and VHDL-AMS for modeling AMS circuits, there is no easy method for generating these models directly from the transistor-level descriptions. This thesis presents an improved method for extracting behavioral models from the simulations of AMS circuits. This method generates labeled Petri net (LPN) models that can be used in the formal verification of circuits, and SystemVerilog models that can be used in the system-level simulations.
Stochastic Control
Uncertainty presents significant challenges in the reasoning about and controlling of complex dynamical systems. To address this challenge, numerous researchers are developing improved methods for stochastic analysis. This book presents a diverse collection of some of the latest research in this important area. In particular, this book gives an overview of some of the theoretical methods and tools for stochastic analysis, and it presents the applications of these methods to problems in systems theory, science, and economics.
State space reductions for scalable verification of asynchronous designs
This paper presents several state space reductions for verifying non-trivial asynchronous designs with a compositional minimization approach. These reductions result in a reduced model that contains the exact set of observably equivalent behavior. Therefore no false counter-examples are produced at the end of verification. The experimental results show good scale-up of compositional minimization using these reductions on a number of asynchronous designs.
Design and analysis of a robust genetic Muller C-element
This paper presents results on the design and analysis of a robust genetic Muller C-element. The Muller C-element is a standard logic gate commonly used to synchronize independent processes in most asynchronous electronic circuits. Synthetic biological logic gates have been previously demonstrated, but there remain many open issues in the design of sequential (state-holding) logic operations. Three designs are considered for the genetic Muller C-element: a majority gate, a toggle switch, and a speed-independent implementation. While the three designs are logically equivalent, each design requires different assumptions to operate correctly. The majority gate design requires the most timing assumptions, the speed-independent design requires the least, and the toggle switch design is a compromise between the two. This paper examines the robustness of these designs as well as the effects of parameter variation using stochastic simulation. The results show that robustness to timing assumptions does not necessarily increase reliability, suggesting that modifications to existing logic design tools are going to be necessary for synthetic biology. Parameter variation simulations yield further insights into the design principles necessary for building robust genetic gates. The results suggest that high gene count, cooperativity of at least two, tight repression, and balanced decay rates are necessary for robust gates. Finally, this paper presents a potential application of the genetic Muller C-element as a quorum-mediated trigger.
iSSA: An incremental stochastic simulation algorithm for genetic circuits
Researchers are now developing synthetic genetic circuits to manipulate the biochemical processes within living cells. In order to model and predict the behavior of these circuits, the designer must account for numerous reactions among many chemical species and genetic components. The analysis of genetic circuits is complicated by the fact that small molecule counts and sporadic gene expression makes stochastic simulation necessary. However, the examination of statistics on ensembles of stochastic simulation runs can hide important behavior. To address this problem, this paper introduces a new method called the incremental stochastic simulation algorithm (iSSA) which determines statistics on typical behavior. This paper illustrates the utility of this algorithm on a circadian rhythm model and a model of a synthetic dual-feedback genetic oscillator.
Automatic abstraction for verification of cyber-physical systems
Models of cyber-physical systems are inherently complex since they must represent hardware, software, and the physical environment. Formal verification of these models is often precluded by state explosion. Fortunately, many important properties may only depend upon a relatively small portion of the system being accurately modeled. This paper presents an automatic abstraction methodology that simplifies the model accordingly. Preliminary results on a fault-tolerant temperature sensor are encouraging.
Analog/mixed-signal circuit verification using models generated from simulation traces
Verification of analog/mixed-signal (AMS) circuits is complicated by the difficulty of obtaining circuit models at suitable levels of abstraction. We propose a method to automatically generate abstract models suitable for formal verification and system-level simulation from transistor-level simulation traces. This paper discusses the application of the proposed methodology to a switched capacitor integrator and PLL phase detector.
Temperature Control of Fimbriation Circuit Switch in Uropathogenic Escherichia coli: Quantitative Analysis via Automated Model Abstraction
Uropathogenic Escherichia coli (UPEC) represent the predominant cause of urinary tract infections (UTIs). A key UPEC molecular virulence mechanism is type 1 fimbriae, whose expression is controlled by the orientation of an invertible chromosomal DNA element—the fim switch. Temperature has been shown to act as a major regulator of fim switching behavior and is overall an important indicator as well as functional feature of many urologic diseases, including UPEC host-pathogen interaction dynamics. Given this panoptic physiological role of temperature during UTI progression and notable empirical challenges to its direct in vivo studies, in silico modeling of corresponding biochemical and biophysical mechanisms essential to UPEC pathogenicity may significantly aid our understanding of the underlying disease processes. However, rigorous computational analysis of biological systems, such as fim switch temperature control circuit, has hereto presented a notoriously demanding problem due to both the substantial complexity of the gene regulatory networks involved as well as their often characteristically discrete and stochastic dynamics. To address these issues, we have developed an approach that enables automated multiscale abstraction of biological system descriptions based on reaction kinetics. Implemented as a computational tool, this method has allowed us to efficiently analyze the modular organization and behavior of the E. coli fimbriation switch circuit at different temperature settings, thus facilitating new insights into this mode of UPEC molecular virulence regulation. In particular, our results suggest that, with respect to its role in shutting down fimbriae expression, the primary function of FimB recombinase may be to effect a controlled down-regulation (rather than increase) of the ON-to-OFF fim switching rate via temperature-dependent suppression of competing dynamics mediated by recombinase FimE. Our computational analysis further implies that this down-regulation mechanism could be particularly significant inside the host environment, thus potentially contributing further understanding toward the development of novel therapeutic approaches to UPEC-caused UTIs.
Abstraction Methods for Analysis of Gene Regulatory Networks
With advances in high throughput methods of data collection for gene regulatory networks, we are now in a position to face the challenge of elucidating how these genes coupled with environmental stimuli orchestrate the regulation of cell-level behaviors. Understanding the behavior of such complex sy…
A New Verification Method For Embedded Systems
Cyber-physical systems, in which computers control real-world mechanisms, are ever more pervasive in our society. These complex systems, containing a mixture of software, digital hardware, and analog circuitry, are often employed in circumstances where their correct behavior is crucial to the safety of their operators. Therefore, verifi cation of such systems would be of great value. This dissertation introduces a modeling and veri fication methodology sufficiently powerful to manage the complications inherent in this mixeddiscipline design space.
Genetic design automation
Electronic design automation (EDA) tools have facilitated the design of ever more complex integrated circuits each year. Synthetic biology would also benefit from the development of genetic design automation (GDA) tools. Existing GDA tools require biologists to design genetic circuits at the molecular level, roughly equivalent to designing electronic circuits at the layout level. Analysis of these circuits is also performed at this very low level. This paper presents the background and issues involved in the development of such a GDA tool for modeling, analysis, and design.
A new verification method for embedded systems
Verification of embedded systems is complicated by the fact that they are composed of digital hardware, analog sensors and actuators, and low level software. In order to verify the interaction of these heterogeneous components, it would be beneficial to have a single modeling formalism that is capable of representing all of these components. To address this need, this paper describes an extended labeled hybrid Petri net (LHPN) model that includes constructs for Boolean, discrete, and continuous variables as well as constructs to model timing. This paper also presents a method to verify these extended LHPNs. Finally, this paper presents a case study to illustrate the application of this model to the verification of a fault-tolerant temperature sensor.
Representing Genetic Networks as Labeled Hybrid Petri Nets for State Space Exploration and Markov Chain Analysis
This paper presents the bachelor’s thesis of Curtis Kendall Madsen which can be broken down into the following three goals. The first goal of this project is to develop a way to convert genetic networks into logical models. Once this is done, finding the state graph of these nets and performing Markov chain analysis on them can provide researchers with insight into the reachability of the states in the original network. Therefore, the second goal of this project is to develop an automated tool that can perform state space exploration of a logical model, and the third goal is to implement a Markov chain analyzer for the stage graph. For the logical representation of genetic networks, the conversion method uses labeled hybrid Petri nets (LHPNs) because they are designed for modeling logic while still allowing for important information required by Markov chain analysis such as transition rates to be included in the model. This conversion method is automated and is integrated into the iBioSim program allowing users to transform a Genetic Circuit Model (GCM) into an LHPN with just a click of a button. Also, iBioSim now includes the LHPN file type in its file tree so users can view and edit LHPNs once conversion is complete. In addition, a method for performing state space exploration on an LHPN allows the user to view the state graph using Graphviz’s Dotty tool.
A Behavioral Synthesis System for Asynchronous Circuits with Bundled-data Implementation
This paper proposes a behavioral synthesis system for asynchronous circuits with bundled-data implementation. The proposed system is based on a behavioral synthesis method for synchronous circuits and extended on operation scheduling and control synthesis for bundled-data implementation. The proposed system synthesizes an RTL model and a simulation model from a behavioral description specified by a restricted C language, a resource library, and a set of design constraints. This paper shows the effectiveness of the proposed system in terms of area and latency through comparisons among bundled-data implementations synthesized by the proposed system, synchronous counterparts, and bundled-data implementations synthesized by using a behavioral synthesis method for synchronous circuits directly.
Verification of Analog/Mixed-Signal Circuits Using Symbolic Methods
This paper presents two symbolic model checking algorithms for the verification of analog/mixed-signal circuits. The first model checker utilizes binary decision diagrams while the second is a bounded model checker that uses a satisfiability modulo theory solver. Both methods have been implemented, and preliminary results are promising.
Efficient Modeling and Verification of Analog/Mixed-Signal Circuits Using Labeled Hybrid Petri Nets
Analog circuit design is traditionally done by expert designers in an ad hoc manner heavily dependent on simulation. This methodology has worked successfully for many years, but process variation and design complexity are prompting designers to explore new techniques. Formal methods are being used successfully to aid in the complex validation problem for digital circuits. This dissertation presents formal methods for analog and mixed-signal (AMS) circuits. This dissertation describes the development of a formal model, labeled hybrid Petri nets (LHPNs), appropriate for the modeling and verification of AMS circuits. An LHPN is a Petri net variant capable of modeling both continuous and discrete quantities. Creating an LHPN model of an AMS circuit by hand is a complicated and error prone exercise that requires expert knowledge. This is unacceptable for practical adoption of the LHPN model and its associated analysis methods. For this reason, this dissertation introduces an automatic LHPN model generation method. The method uses a set of simulation traces and a desired system property to generate an LHPN modeling the behavior of the simulation traces. The model generator can also be used to generate abstract Verilog-AMS or VHDL-AMS models suitable for use in system-level simulations. Formal verification of a property over the entire state space of an LHPN model is complicated by the infinite state of the model. For this reason, the infinite states of the model are grouped into potentially finite groups of equivalent states for verification. Difference bound matrices (DBMs), a restricted form of convex polygons, are used to represent these equivalent classes of infinite states. Reachability analysis using DBMs is very efficient at the cost of exactness. This dissertation presents algorithms for conservative state space analysis and verification of LHPNs. Finally, these methods are demonstrated on several case studies of AMS circuits from both academia and industry. The formal verification methods demonstrate the ability to find bugs missed by standard simulations. The abstract modeling methods show the promise of using automatically generated abstract models by demonstrating up to 40x speedup for some examples.
Production-passage-time approximation: a new approximation method to accelerate the simulation process of enzymatic reactions
Given the substantial computational requirements of stochastic simulation, approximation is essential for efficient analysis of any realistic biochemical system. This paper introduces a new approximation method to reduce the computational cost of stochastic simulations of an enzymatic reaction scheme which in biochemical systems often includes rapidly changing fast reactions with enzyme and enzyme-substrate complex molecules present in very small counts. Our new method removes the substrate dissociation reaction by approximating the passage time of the formation of each enzyme-substrate complex molecule which is destined to a production reaction. This approach skips the firings of unimportant yet expensive reaction events, resulting in a substantial acceleration in the stochastic simulations of enzymatic reactions. Additionally, since all the parameters used in our new approach can be derived by the Michaelis-Menten parameters which can actually be measured from experimental data, applications of this approximation can be practical even without having full knowledge of the underlying enzymatic reaction. Here, we apply this new method to various enzymatic reaction systems, resulting in a speedup of orders of magnitude in temporal behavior analysis without any significant loss in accuracy. Furthermore, we show that our new method can perform better than some of the best existing approximation methods for enzymatic reactions in terms of accuracy and efficiency.
Design and Analysis of Genetic Circuits
Electronic Design Automation (EDA) tools have facilitated the design of ever more complex integrated circuits each year. Synthetic biology would also benefit from the development of Genetic Design Automation (GDA) tools. Existing GDA tools require biologists to design genetic circuits at the molecular level, roughly equivalent to designing electronic circuits at the layout level. Analysis of these circuits is also performed at this very low level. This thesis presents a first step at developing a GDA tool that supports higher levels of abstraction. In particular, this thesis describes the Genetic Circuit Model (GCM), a graphical specification language from which molecular descriptions can be synthesized. The GCM has several advantages. The input is tightly controlled through the use of an editor, limiting the possibility of user error. The representation of the genetic circuit is much more compact than using the System Biology Markup Language (SBML), the standard form for representing genetics circuits. The GCM can be automatically translated into SBML, allowing GCM’s to be easily simulated across multiple different simulators. The GCM to SBML translation process is targeted in such a way that the resulting output can be easily abstracted to allow for efficient simulation. To evaluate and test the GCM, this thesis presents a case study of the design of a genetic Muller C-element, a gate often used in asynchronous design. Three different genetic Muller C-elements are designed and analyzed. The utility of the GCM is demonstrated as it allows for efficient analysis of the Muller C-elements. The results of the simulations show that logically equivalent circuits can have different behaviors. In particular, a speed independent Muller C-element does not necessary imply that the gate is more robust than a non-speed independent gate. Design principles gathered from the simulations are that dual-rail outputs are essential, high gene count increases robustness, cooperativity greater than one is necessary, repression needs to be strong, and decay rates must be balanced for high robustness and low switching time. One potential application of the genetic Muller C-element is determining when to start the invasion of cancer cells. The two input signals are an environmental signal, and a communication signal. Using these signals, the bacteria colony can correctly reach consensus on when to begin the invasion. One interesting result is that noise is necessary in correctly switching into the invasion state.
Abstract Modeling and Simulation Aided Verification of Analog/Mixed-Signal Circuits
Abstract. Analog/Mixed-signal (AMS) circuit verification is a growing problem as process variation increases and AMS circuits become more functionally complex. To improve analog verification flows, AMS circuit models are needed at different levels of abstraction. This paper discusses recent work and future directions for abstract model generation and simulation aided verification of AMS circuits. In particular, a CMOS ring oscillator with feedforward inverters is used as a motivating example for the work. This example highlights progress and future directions in AMS modeling and verification. 1
Synthesis of Genetic Circuits from Graphical Specifications
EDA tools have facilitated the design of ever more complex integrated circuits each year. Synthetic biology would also benefit from the development of genetic design automation (GDA) tools. Existing GDA tools require biologists to design genetic circuits at the molecular level, roughly equivalent to designing electronic circuits at the layout level. Analysis of these circuits is also performed at this very low level. This paper presents a first step at developing a GDA tool that supports higher levels of abstraction. In particular, this paper describes a graphical specification language from which molecular descriptions can be synthesized. These descriptions can then be abstracted to facilitate efficient analysis. I.
A behavioral synthesis method for asynchronous circuits with bundled-data implementation (Tool paper)
This paper presents a behavioral synthesis method for asynchronous circuits with bundled-data implementation. This paper extends a behavioral synthesis method for synchronous circuits so that an RTL model of bundled-data implementation is synthesized from a behavioral description specified by a restricted C language. Finally, this paper evaluates our method for several benchmarks through a tool implementation.
Model Abstraction and Temporal Behavior Analysis of Genetic Regulatory Networks
With advances in technologies such as high throughput data collection and genome sequencing methods, the Human Genome Project which, among other things, determined the human DNA sequence and identified all the genes in human DNA was completed at least two years ahead of time. As these technologies are becoming more accurate, efficient, and cost effective and a massive amount of genomic and proteomic data are becoming available at a rapid pace, we are now in the position to face the challenge to understand how these genes coupled with environmental stimuli orchestrate the regulation of cell-level behaviors. However, understanding such complex systems is very expensive and is most likely impractical with wet-lab experiments alone as the amount and the complexity of data substantially increase, requiring the integration of computational methods to make the process more efficient. To allow for substantial acceleration in computational analysis, this dissertation develops a model abstraction methodology for biochemical systems which systematically performs various model abstractions to reduce the complexity of computational biochemical models. Our methodology is particularly useful for systems with small molecular counts that require the discrete and stochastic representation and thus demand substantial computational requirements. As a case study, this dissertation illustrates the application of individual abstraction methods to such systems. Furthermore, it demonstrates the application of collective abstraction methods at various accuracy levels to temporal behavior analysis of several genetic regulatory networks. This dissertation shows that analysis time of biologically relevant properties of such genetic regulatory networks can be improved from days of work to minutes of work using our methodology while maintaining reasonable accuracy.
Learning Genetic Regulatory Network Connectivity from Time Series Data
Recent experimental advances facilitate the collection of time series data that indicate which genes in a cell are expressed. This information can be used to understand the genetic regulatory network that generates the data. Typically, Bayesian analysis approaches are applied which neglect the time series nature of the experimental data, have difficulty in determining the direction of causality, and do not perform well on networks with tight feedback. To address these problems, this dissertation presents an improved method, called the GeneNet algorithm, to learn genetic regulatory network connectivity which exploits the time series nature of experimental data to allow for better causal predictions on networks with tight feedback. More specifically, the GeneNet algorithm provides several contributions to the area of genetic network discovery. It finds networks with cyclic or tight feedback behavior often missed by other methods as it performs a more local analysis of the data. It provides the researcher with the ability to see the interactions between genes in a genetic network. It guides experimental design by providing feedback to the researcher as to which parts of the network are the most unclear. It is encased in an infrastructure that allows for rapid genetic network model creation and evaluation. The GeneNet algorithm first encodes the data into levels. Next, it determines an initial set of influence vectors for each species based upon the probability of the species’ expression increasing. From this set of influence vectors, it determines if any influence vectors should be merged, representing a combined effect. Finally, influence vectors are competed against each other to obtain the best influence vector. The result is a directed graph representation of the genetic network’s repression and activation connections. Results are reported for several synthetic networks showing significant improvements in both recall and runtime while performing nearly as well or better in precision over a dynamic Bayesian approach.
Application of Automated Model Generation Techniques to Analog/Mixed-Signal Circuits
Abstract models of analog/mixed-signal (AMS) circuits can be used for formal verification and system-level simulation. The difficulty of creating these models precludes their widespread use. This paper presents an automated method to generate abstract models appropriate for system-level simulation and formal verification. This method uses simulation traces and thresholds on the design variables to generate a piecewise-linear representation of the system. This piecewise-linear representation can be converted to a Verilog-AMS model or a Labeled Hybrid Petri Net formal model. Results are presented for the model generation, simulation, and verification of a PLL phase detector circuit.
Bounded Model Checking of Analog and Mixed-Signal Circuits Using an SMT Solver
This paper presents a bounded model checking algorithm for the verification of analog and mixed-signal (AMS) circuits using a satisfiability modulo theories (SMT) solver. The systems are modeled in VHDL-AMS, a hardware description language for AMS circuits. In this model, system safety properties are specified as assertion statements. The VHDL-AMS description is compiled into labeled hybrid Petri nets (LHPNs) in which analog values are modeled as continuous variables that can change at rates in a bounded range and digital values are modeled using Boolean signals. The verification method begins by transforming the LHPN model into an SMT formula composed of the initial state, the transition relation unrolled for a specified number of iterations, and the complement of the assertion in each set of state variables. When this formula evaluates to true, this indicates a violation of the assertion and an error trace is reported. This method has been implemented and preliminary results are promising.
Analog/Mixed-Signal Circuit Verification Using Models Generated from Simulation Traces
Formal and semi-formal verification of analog/mixed-signal circuits is complicated by the difficulty of obtaining circuit models suitable for analysis. We propose a method to generate a formal model from simulation traces. The resulting model is conservative in that it includes all of the original simulation traces used to generate it plus additional behavior. Information obtained during the model generation process can also be used to refine the simulation and verification process.
Verification of Analog and Mixed-Signal Circuits Using Symbolic Methods
With the rapidly increasing complexity of hardware, traditional validation techniques are becoming insufficient. This has led to a substantial interest in the formal verification of digital components. There has been relatively little research, however, into the application of formal verification methods to the analog/mixed-signal domain. Therefore, the overall goal of this work is to provide a system for efficient and meaningful analysis of analog/mixed-signal circuits. This encompasses two major efforts: modeling and symbolic analysis. The continuous nature of analog circuits requires a modeling method that is capable of representing continuous behavior and the discrete nature of digital circuits requires a modeling method that is capable of representing discrete behavior. This dual requirement necessitates a hybrid model—a model that can simultaneously represent continuous and discrete behavior. This work details the development of a specialized hybrid Petri net model with capabilities similar to hybrid automata. Analysis is greatly complicated by the addition of continuous behavior to the model. To help alleviate this, infinite numbers of states are often grouped into equivalence classes represented by symbolic structures. The analysis methods described here represent ranges of continuous variables using groups of inequalities which are then either mapped to Binary Decision Diagram variables so that necessary operations can be performed efficiently, or handed over to an advanced Satisfiability Modulo Theories solver for analysis. After describing the verification system in detail, experiences applying the techniques to several case studies are described and performance results are provided.
Synthesis of Timed Circuits Based on Decomposition
This paper presents a decomposition-based method for timed circuit design that is capable of significantly reducing the cost of synthesis. In particular, this method synthesizes each output individually. It begins by contracting the timed signal transition graph (STG) to include only transitions on the output of interest and its possible trigger signals. Next, the reachable state space for this contracted STG is analyzed to determine a minimal number of additional signals, which must be reintroduced into the STG to obtain complete state coding. The circuit for this output is then synthesized from this STG. Results show that the quality of the circuit implementation is nearly as good as the one found from the full reachable state space, but it can be applied to find circuits for which full-state-space methods cannot be successfully applied. The proposed method has been implemented as a part of our tool Nii-Utah Timed Asynchronous circuit Synthesis system (nutas), and its first version is available at http://research.nii.ac.jp/ yoneda.
Hazard Checking of Timed Asynchronous Circuits Revisited
This paper proposes a new approach for the hazard checking of timed asynchronous circuits. Previous papers proposed either exact algorithms, which suffer from statespace explosion, or efficient algorithms which use a (conservative) approximation to avoid state-space explosion but can result in the rejection of designs which are valid. In particular, [7] presents a timed extention of the work in [1] which is very efficient but is not able to handle circuits with internal loops, which prevents its use in some cases. We propose a new approach to the problem in order to overcome the mentioned limitations, without sacrificing efficiency. To do so, we first introduce a general framework targeted at the conservative checking of safety failures. This framework is not restricted to the checking of timed asynchronous circuits. Secondly, we propose a new (conservative) semantics for timed circuits, in order to use the proposed framework for hazard checking of such circuits. Using this framework with the proposed semantics yields an efficient algorithm that addresses the limitations of the previous approaches.
Production-Passage-Time Approximation: A New Approximation Method to Accelerate the Simulation Process of Enzymatic Reactions
Given the substantial computational requirements of stochastic simulation, approximation is essential for efficient analysis of any realistic biochemical system. This paper introduces a new approximation method to reduce the computational cost of stochastic simulations of an enzymatic reaction scheme which in biochemical systems often includes rapidly changing fast reactions with enzyme and enzyme-substrate complex molecules present in very small counts. Our new method removes the substrate dissociation reaction by approximating the passage time of the formation of each enzyme-substrate complex molecule which is destined to a production reaction. This approach skips the firings of unimportant yet expensive reaction events, resulting in a substantial acceleration in the stochastic simulations of enzymatic reactions. Additionally, since all the parameters used in our new approach can be derived by the Michaelis-Menten parameters which can actually be measured from experimental data, applications of this approximation can be practical even without having full knowledge of the underlying enzymatic reaction. Furthermore, since our approach does not require a customized simulation procedure for enzymatic reactions, it allows biochemical systems that include such reactions to still take advantage of standard stochastic simulation tools. Here, we apply this new method to various enzymatic reaction systems, resulting in a speedup of orders of magnitude in temporal behavior analysis without any significant loss in accuracy.
The Design of a Genetic Muller C-Element
Synthetic biology uses engineering principles to design circuits out of genetic materials that are inserted into bacteria to perform various tasks. While synthetic combinational Boolean logic gates have been constructed, there are many open issues in the design of sequential logic gates. One such gate common in most asynchronous circuits is the Muller C-element, which is used to synchronize multiple independent processes. This paper proposes a novel design for a genetic Muller C-element using transcriptional regulatory elements. The design of a genetic Muller C-element enables the construction of virtually any asynchronous circuit from genetic material. There are, however, many issues that complicate designs with genetic materials. These complications result in modifications being required to the normal digital design procedure. This paper presents two designs that are logically equivalent to a Muller C-element. Mathematical analysis and stochastic simulation, however, show that only one functions reliably.
Efficient Verification of Hazard-Freedom in Gate-Level Timed Asynchronous Circuits
This paper presents an efficient method for verifying hazard-freedom in gate-level timed asynchronous circuits. Timed circuits are a class of asynchronous circuits that are optimized using explicit timing information. In asynchronous circuits, correct operation requires that there are no hazards in the circuit implementation. Therefore, when designing an asynchronous circuit, each internal node and output of the circuit must be verified for hazard-freedom to ensure correct operation. Current verification algorithms for timed circuits require an explicit state exploration that often results in state explosion for even modest-sized examples. The goal of this paper is to abstract the behavior of internal nodes and utilize this information to make a conservative determination of hazard-freedom for each node in the circuit. Experimental results indicate that this approach is substantially more efficient than existing timing verification tools. These results also indicate that this method scales well for large examples that could not be previously analyzed, in that it is capable of analyzing these circuits in less than a second. While this method is conservative in that some false hazards may be reported, our results indicate that their number is small
Symbolic Model Checking of Analog/Mixed-Signal Circuits
This paper presents a Boolean based symbolic model checking algorithm for the verification of analog/mixed-signal (AMS) circuits. The systems are modeled in VHDL-AMS, a hardware description language for AMS circuits. The VHDL-AMS description is compiled into labeled hybrid Petri nets (LH-PNs) in which analog values are modeled as continuous variables that can change at rates in a bounded range and digital values are modeled using Boolean signals. System properties are specified as temporal logic formulas using timed CTL (TCTL). The verification proceeds over the structure of the formula and maps separation predicates to Boolean variables. The state space is thus represented as a Boolean function using a binary decision diagram (BDD) and the verification algorithm relies on the efficient use of BDD operations.
Verification of Analog/Mixed-Signal Circuits Using Labeled Hybrid Petri Nets
System on a chip design results in the integration of digital, analog, and mixed-signal circuits on the same substrate which further complicates the already difficult validation problem. This paper presents a new model, labeled hybrid Petri nets (LHPNs), that is developed to be capable of modeling such a heterogeneous set of components. This paper also describes a compiler from VHDL-AMS to LHPNs. To support formal verification, this paper presents an efficient zone-based state space exploration algorithm for LHPNs. This algorithm uses a process known as warping to allow zones to describe continuous variables that may be changing at variable rates. Finally, this paper describes the application of this algorithm to a couple of analog/mixed-signal circuit examples
ILP-based Scheduling for Asynchronous Circuits in Bundled-Data Implementation
In this paper, we propose a new scheduling method for asynchronous circuits in bundled-data implementation. The method is based on integer linear programming (ILP) which explores an optimum schedule under resource or time constraints. To schedule descriptions with many operations, our method approximate start times of operations and formulate an ILP based on the approximated start times. Because less numbers of variables and constraints are required compared to the traditional ILP formulation, the schedule of operations is determined in short time preserving the quality of resulting circuit.
The Case for Analog Circuit Verification
The traditional approach to validate analog circuits is to utilize extensive SPICE-level simulations. The main challenge of this approach is knowing when all important corner cases have been simulated. A new alternative is to utilize formal verification techniques. This paper utilizes a simple example to illustrate the potential flaws of a simulation-only based validation methodology and the potential benefits of formal verification of analog circuits.
Abstracted stochastic analysis of type 1 pili expression in E. coli
Abstract — With the aid of model abstractions, biochemical networks can be analyzed at different levels of resolution: from low-level quantitative models to high-level qualitative ones. Furthermore, an ability to change the level of abstraction can be very useful when dealing with many biological systems, including gene regulatory networks. These systems typically have too many components and states to be practically studied using all-inclusive low-level models, yet they often manifest enough dynamical and functional complexity, making an entirely high-level qualitative representation similarly inadequate — thus necessitating a search for some intermediate level of abstraction. Finally, while most abstractions used in modeling of biochemical networks have traditionally been performed manually, doing so accurately in a large system is a tedious and time-consuming process that is highly susceptible to errors during model transformation. To address these issues, we have developed a methodology and implemented an automated modeling and analysis tool with variable abstraction level capabilities. In this paper, we use it for the analysis of switching in Type 1 pili expression dynamics and, in particular, for the problem of estimating the effect of H-NS and Lrp regulatory protein levels on phase variation rates in E. coli. Such behavior is notoriously difficult to study due to the size of the associated gene regulatory network and the characteristically stochastic dynamics involved, which result in very high analytical and computational demands. Here, we show how, by using our system, we are able to automatically abstract the switch network and accurately predict E. coli afimbriation rates, while, at the same time, accelerating the required computations by up to two orders of magnitude. I.
Verification of timed circuits with failure-directed abstractions
This paper presents a method to address state explosion in timed-circuit verification by using abstraction directed by the failure model. This method allows us to decompose the verification problem into a set of subproblems, each of which proves that a specific failure condition does not occur. To each subproblem, abstraction is applied using safe transformations to reduce the complexity of verification. The abstraction preserves all essential behaviors conservatively for the specific failure model in the concrete description. Therefore, no violations of the given failure model are missed when only the abstract description is analyzed. An algorithm is also shown to examine the abstract error trace to either find a concrete error trace or report that it is a false negative. This paper presents results using the proposed failure-directed abstractions as applied to several large timed-circuit designs.
Learning genetic regulatory network connectivity from time series data
Abstract. Recent experimental advances facilitate the collection of time series data that indicate which genes in a cell are expressed. This paper proposes an efficient method to generate the genetic regulatory network inferred from time series data. Our method first encodes the data into levels. Next, it determines the set of potential parents for each gene based upon the probability of the gene’s expression increasing. After a subset of potential parents are selected, it determines if any genes in this set may have a combined effect. Finally, the potential sets of parents are competed against each other to determine the final set of parents. The result is a directed graph representation of the genetic network’s repression and activation connections. Our results on synthetic data generated from models for several genetic networks with tight feedback are promising. 1
Failure Trace Analysis of Timed Circuits for Automatic Timing Constraints Derivation
This work proposes a technique to automatically obtain timing constraints for a given timed circuit to operate correctly. A designated set of delay parameters of a circuit are first set to sufficiently large bounds, and verification runs followed by failure analysis are repeated. Each verification run performs timed state space enumeration under the given delay bounds, and produces a failure trace if it exists. The failure trace is analyzed, and sufficient timing constraints to prevent the failure are obtained. Then, the delay bounds are tightened according to the timing constraints by using an ILP (Integer Linear Programming) solver. This process terminates when either some delay bounds under which no failure is detected are found or no new delay bounds to prevent the failures can be obtained. The experimental results using a naive implementation show that the proposed method can efficiently handle asynchronous benchmark circuits and nontrivial GasP circuits.
Partial Order Reduction for Detecting Safety and Timing Failures of Timed Circuits
This paper proposes a partial order reduction algorithm for timed trace theoretic verification in order to detect both safety failures and timing failures of timed circuits efficiently. This algorithm is based on the framework of timed trace theoretic verification according to the original untimed trace theory. Consequently, its conformance checking supports hierarchical structure when verifying timed circuits. Experimenting with the STARI and DME circuits, the proposed approach shows its effectiveness.
Asynchronous abstraction methodology for genetic regulatory networks
Abstract. In order to efficiently analyze a large scale system in an automated and objective manner, abstraction is essential. This paper presents an automated abstraction methodology that systematically reduces the small scale complexity found in genetic regulatory network models, while broadly preserving the large scale system behavior. Our approach is to first reduce the number of reactions through a quasisteady-state approximation-based algorithm. Second, it represents the exact molecular state of the system by a set of reduced Boolean (or nary) discrete levels. This results in a chemical master equation that is approximated by a Markov chain with a much smaller state space providing significant simulation time acceleration and computability gains. 1 Background Numerous methods have been proposed to model genetic regulatory networks [1, 2]. While many traditional approaches have relied on a differential equation representation as inferred from a set of underlying biochemical reactions, there has been a growing appreciation of their limitations [3-6]. In particular, differential equation analysis of genetic networks generally assumes that the number of molecules in a cell is high and their concentrations can be viewed as continuous quantities, while their underlying reactions occur deterministically. However, in natural genetic networks these assumptions frequently do not hold as, for example, DNA molecules are typically present in single digit quantities, while some promoters can lead to substantial fluctuations in transcription/translation rates and essentially non-deterministic expression characteristics [7, 8].
High level synthesis of timed asynchronous circuits
This paper proposes applying a logic synthesis approach to high level synthesis from SpecC specifications to timed asynchronous gate-level circuits. The state-based logic synthesis is used to allow for global and timing optimization. In order to reduce the overhead in resetting phases, a protocol called early acknowledgment protocol and its STG (signal transition graph) generation technique are proposed. In this protocol, the state variables inserted to guarantee that STGs have CSC (complete state coding) usually cause no overhead. The experiments to synthesize a portion of a DCT circuit show that the proposed method can handle a nontrivial example and produce a smaller and faster circuit than a previous approach.
Technology Mapping of Timed Asynchronous Circuits
This dissertation presents an efficient method for technology-mapping of timedasynchronous circuits. Technology-mapping combines the steps of decomposition, partitioning, and matching/covering to implement a synthesized design in a given technology. This work is applied to timed circuits, which are a class of asynchronous circuits that utilize explicit timing information for optimization throughout the entire design process. This work carries the timing constraints down to the circuit implementation level, giving new insight into the detection and elimination of hazards. In asynchronous circuits, correct operation requires that there are no hazards in the circuit implementation. Therefore, each internal node and output of the transformed circuit following decomposition must be verified for hazard-freedom to ensure correct operation. Current verification algorithms require an explicit state exploration often resulting in state explosion for even modest sized examples. The goal of the hazard verification portion of technology-mapping is to abstract the behavior of internal nodes and utilize the reduced state space to make a conservative determination of hazard-freedom for each node in the circuit. The newly annotated circuit is then mapped to an existing library for implementation. This dissertation explores various complexities of libraries used for matching and examines the hazard covering behavior using a variety of gates. Issues such as short-circuit detection and common-input matching are explored in detail, particularly when libraries contain generalized C-elements. The goal of this research is a hazard-free implementation of the synthesized design in a user-provided technology. Experimental results indicate that this new hazard-verification approach is substantially more efficient than existing timing verification tools and that in most cases hazard-free netlists are produced with modest sized libraries.
Verification of Analog and Mixed-Signal Circuits Using Timed Hybrid Petri Nets
Embedded systems are composed of a heterogeneous collection of digital, analog, and mixed-signal hardware components. This paper presents a method for the verification of systems composed of such a variety of components. This method utilizes a new model, timed hybrid Petri nets (THPN), to model these circuits. In particular, this paper describes an efficient, approximate algorithm to find the reachable states of a THPN model. Using this state space, desired properties specified in ACTL are verified. To demonstrate these methodologies, a few hybrid automata benchmarks, a tunnel diode oscillator, and a phase-locked loop are modeled and analyzed using THPNs.
Partial Order Reduction for Detecting Safety and Timing Failures of Timed Circuits
This paper proposes a partial order reduction algorithm for timed trace theoretic verification in order to detect both safety failures and timing failures of timed circuits efficiently. This algorithm is based on the framework of timed trace theoretic verification according to the original untimed trace theory. Consequently, its conformance checking supports hierarchical verification. Experimenting with the STARI circuits, the proposed approach shows its effectiveness.
Synthesis of speed independent circuits based on decomposition
This work presents a decomposition method for speed-independent circuit design that is capable of significantly reducing the cost of synthesis. In particular, this method synthesizes each output individually. It begins by contracting the STG to include only transitions on the output of interest and its trigger signals. Next, the reachable state space for this contracted STG is analyzed to determine a minimal number of additional signals which must be reintroduced into the STG to obtain CSC. The circuit for this output is then synthesized from this STG. Results show that the quality of the circuit implementation is nearly as good as the one found from the full reachable state space, but it can be applied to find circuits for which full state space methods cannot be successfully applied. The proposed method has been implemented as a part of our tool nutas (Nii-Utah timed asynchronous circuit synthesis system).
CMOS analog MAP decoder for (8,4) Hamming code
Design and test results for a fully integrated translinear tail-biting MAP error-control decoder are presented. Decoder designs have been reported for various applications which make use of analog computation, mostly for Viterbi-style decoders. MAP decoders are more complex, and are necessary components of powerful iterative decoding systems such as turbo codes. Analog circuits may require less area and power than digital implementations in high-speed iterative applications. Our (8, 4) Hamming decoder, implemented in an AMI 0.5-/spl mu/m process, is the first functioning CMOS analog MAP decoder. While designed to operate in subthreshold, the decoder also functions above threshold with a small performance penalty. The chip has been tested at bit rates up to 2 Mb/s, and simulations indicate a top speed of about 10 Mb/s in strong inversion. The decoder circuit size is 0.82 mm/sup 2/, and typical power consumption is 1 mW at 1 Mb/s.
Partial Order Reduction for Timed Circuit Verification Based on Level Oriented Model
Using a level oriented model for verification of asynchronous circuits helps users to easily construct formal models with high readability or to naturally model data-path circuits. On the other hand, in order to use such a model for larger circuit, some technique to avoid the state explosion problem is essential. This paper first defines a level oriented formal model based on time Petri nets, and then proposes a partial order reduction algorithm that prunes unnecessary state generation while guaranteeing the correctness of the verification.
Efficient verification of hazard-freedom in gate-level timed asynchronous circuits
This paper presents an efficient method for verifying hazard freedom in timed asynchronous circuits. Timed circuits are a class of asynchronous circuits that utilize explicit timing information for optimization throughout the entire design process. In asynchronous circuits, correct operation requires that there are no hazards in the circuit implementation. Therefore, when designing an asynchronous circuit, each internal node and output of the circuit must be verified for hazard-freedom to ensure correct operation. Current verification algorithms for timed asynchronous circuits require an explicit state exploration often resulting in state explosion for even modest sized examples. The goal of this work is to abstract the behavior of internal nodes and utilize this information to make a conservative determination of hazard-freedom for each node in the circuit. Experimental results indicate that this approach is substantially more efficient than existing timing verification tools. These results also indicate that this method scales well for large examples. It is capable of analyzing circuits in less than a second that could not be previously analyzed. While this method is conservative in that some false hazards may be reported, our results indicate that the number of false hazards is small.
Modular verification of timed circuits using automatic abstraction
The major barrier that prevents the application of formal verification to large designs is state explosion. This paper presents a new approach for verification of timed circuits using automatic abstraction. This approach partitions the design into modules, each with constrained complexity. Before verification is applied to each individual module, irrelevant information to the behavior of the selected module is abstracted away. This approach converts a verification problem with big exponential complexity to a set of subproblems, each with small exponential complexity. Experimental results are promising in that they indicate that our approach has the potential of completing much faster while using less memory than traditional flat analysis.
A Comparison of Timed State Space Analysis Methods
Circuit designers continue to push the limits of high speed circuit design. This desire for speed motivates the creation of new and innovative design styles. One of these design styles is timed circuits. Timed circuits take advantage of timing information to increase performance. This style has been applied in industrial research to designs like IBM’s guTS microprocessor, the Intel RAPPID project, and Sun’s GasP circuits. These experimental designs were successful at increasing performance, but they are only experimental designs. Timed circuits have not yet been used in a commercial design. One problem that plagues timed circuit design is the difficulty of understanding the complex timing interactions between circuit components. In response to this problem, researchers have created several methods and tools to help verify timed circuit designs. One of the most critical aspects of these methods is state space exploration of both the timed and untimed state space. This work concentrates on two leading methods to do timed state space exploration using Petri nets. These methods were previously implemented in two different CAD tools, ATACS and VINAS-P. However, a clear comparison of the methods has not been done due to the fact that the methods were implemented in different tools. We extended the ATACS framework by adding the methods previously only used by VINAS-P. Having both methods implemented in the same tool allows us to do an accurate comparison of the methods to better understand the strengths and weaknesses of each method.
Modular Synthesis of Timed Circuits Using Partial Order Reduction
This paper develops a modular synthesis algorithm for timed circuits that is dramatically accelerated by partial order reduction. This algorithm synthesizes each module in a hierarchical design individually. It utilizes partial order reduction to reduce the state space explored for the other modules by considering a single interleaving of concurrently enabled transitions. This approach better manages the state explosion problem resulting in a more than 2 order of magnitude reduction in synthesis time. The improved synthesis time enables the synthesis of a larger class of timed circuits than was previously possible.
Level oriented formal model for asynchronous circuit verification and its efficient analysis method
Using a level-oriented model for verification of asynchronous circuits helps users to easily construct formal models with high readability or to naturally model datapath circuits. On the other hand, in order to use such a model on large circuits, techniques to avoid the state explosion problem must be developed. This paper first introduces a level-oriented formal model based on time Petri nets, and then proposes its partial order reduction algorithm that prunes unnecessary state generation while guaranteeing the correctness of the verification.
Design Methodology for Analog VLSI Implementations of Error Control Decoders
In order to reach the Shannon limit, researchers have found more efficient error control coding schemes. However, the computational complexity of such error control coding schemes is a barrier to implementing them. Recently, researchers have found that bioinspired analog network decoding is a good approach with better combined power/speed performance than its digital counterparts. However, the lack of CAD (computer aided design) tools makes the analog implementation quite time consuming and error prone. Meanwhile, the performance loss due to the nonidealities of the analog circuits has not been systematically analyzed. Also, how to organize analog circuits so that the nonideal effects are minimized has not been discussed. In designing analog error control decoders, simulation is a time-consuming task because the bit error rate is quite low at high SNR (signal to noise ratio), requiring a large number of simulations. By using high-level VHDL simulations, the simulation is done both accurately and efficiently. Many researchers have found that error control decoders can be interpreted as operations of the sum-product algorithm on probability propagation networks, which is a kind of factor graph. Of course, analog error control decoders can also be described at a high-level using factor graphs. As a result, an automatic simulation tool is built. From its high-level factor graph description, the VHDL simulation files for an analog error control decoder can be automatically generated, making the simulation process simple and efficient. After analyzing the factor graph representations of analog error control decoders, we found that analog error control decoders have quite regular structures and can be built by using a small number of basic cells in a cell library, facilitating automatic synthesis. This dissertation also presents the cell library and how to automatically synthesize analog decoders from a factor graph description. All substantial nonideal effects of the analog circuit are also discussed in the dissertation. How to organize the circuit to minimize these effects and make the circuit optimized in a combined consideration of speed, performance, and power is also provided.
Correctness and Reduction in Timed Circuit Analysis
To increase performance, circuit designers are experimenting with timed circuits a class of circuits that rely on a complex set of timing constraints for correct functionality. This is evidenced in published experimental designs from industry. Timing constraints are key to the success of these designs, and algorithms to verify timing constraints are required to make them practical in commercial applications. Due to the complexity of the constraints, however, traditional static timing analysis is not adequate. Timed state space analysis is required; thus, improved timed state space analysis is paramount to producing efficient timed circuits. This dissertation discusses two facets of work in timed state space analysis: correctness and reduction. For correctness, this dissertation presents the level ruled Petri net as a model for timed circuits. This model is based on the Petri net language. It includes, however, timing information and level expressions that are key to the specification and verification of timed circuits. This dissertation formalizes the intent of correctness in the verification of a timed circuit by defining a set of failure conditions that can be analyzed in the circuit’s respective model. The circuit is said to be correct if its model is failure free. For reduction, this dissertation presents a timed state space analysis algorithm that verifies correctness in the timed circuit model. The algorithm, when compared to existing algorithms, reduces on average the running time and memory footprint of analysis. A partial order reduction is implemented for the algorithm to further reduce its resource usage. This reduction is not supported by the existing algorithms; thus, the new analysis algorithm can be applied to systems that are beyond their capacity. This is demonstrated in verifying industrial designs from IBM and Sun Microsystems.
Complete State Coding of Timed Asynchronous Circuits
This thesis describes a method or solving the complete state coding problem for timed asynchronous systems in an efficient manner. Timed asynchronous systems differ from untimed, speed independent systems in that any change to the system or its timing may dramatically affect the reachable state space. Because frequent state space exploration is time consuming, timing information is used in a variety of ways to postpone or eliminate state space explorations. First, timing information is used to predict the impact of a state signal on the overall system. Second, concurrency information is used to narrow the search space to timing-unique solutions. Third, timing information allows state signal insertion in a timing-sequential, yet noncausal, manner. This permits insertion before input events, an option not readily available in speed-independent systems. Finally, by considering timing, state signal insertion points can be chosen which minimally increase circuit latency. The method has been implemented in the automated design tool ATACS, and correctly and efficiently completes the state code for a variety of established state coding benchmark systems.
Efficient algorithms for exact two-level hazard-free logic minimization
This paper presents a new approach to two-level hazard-free logic minimization in the context of extended burst-mode finite-state machine synthesis. The approach achieves fast single-output logic minimization that yields solutions that are exact in the number of literals. This paper presents algorithms and hazard constraints targeting both generalized C-element and two-level standard gate implementations. The logic minimization approach presented in this paper is based on state graph exploration in conjunction with single-cube cover algorithms. The algorithm achieves fast logic minimization by using compacted state graphs, cover tables, and a divide-and-merge algorithm for efficient single output minimization. The exact two-level hazard-free logic minimizer presented in this paper finds a minimal number of literal solutions and is several orders of magnitude faster than existing literal exact methods for the largest benchmarks available to date. This includes a benchmark that has never been possible to solve exactly in number of literals before.
Framework of Timed Trace Theoretic Verification Revisited
This paper develops a framework to support trace theoretic verification of timed circuits and systems. A theoretical foundation for classifying timed traces as either successes or failures is developed. The concept of the semimirror is introduced to allow conformance checking thus supporting hierarchical verification of timed circuits and systems. Finally, we relate our framework to those previously proposed for timing verification.
Protocol Selection, Implementation, and Analysis for Asynchronous Circuits
This dissertation presents new methods for handshaking expansion of asynchronous circuits. Handshaking expansion includes protocol selection, reshuffling, and state variable insertion. The starting point is a channel-level specification of a design. The goal is a signal-level description of the given design that is correct, synthesizable, and efficient. This dissertation studies the impact of protocol selection and implementation on deadlock avoidance, complete state coding (CSC), CPU time required to compile a given example, and the quality of the circuit. This dissertation treats reshuffling and state-variable insertion as special cases of concurrency reduction. Prior work in the field has also taken this approach. However, this dissertation extends this approach and applies it to specifications that contain quantitative timing assumptions. The concurrency reduction algorithms have been implemented within a computer aided design (CAD) tool. Starting from a signal-level specification that contains the constraints of the desired protocol, these algorithms search the concurrency reduction design space, guided by an estimate of the performance of the final circuit. The CAD tool that this dissertation presents also contains a front end that, given a channel-level specification, produces the starting point for concurrency-reduction. This front end currently handles only pure synchronization channels, using one protocol. Finding all possible ways to reduce concurrency of a specification is a fundamentally exponential problem. However, this dissertation presents techniques to dramatically prune the search space. This dissertation demonstrates that these techniques are capable of reducing the search space by several orders of magnitude compared to the theoretical upper bound – and by one order of magnitude beyond existing techniques – without significantly impacting the quality of the solutions.
Automatic Derivation of Timing Constraints by Failure Analysis
This work proposes a technique to automatically obtain timing constraints for a given timed circuit to operate correctly. A designated set of delay parameters of a circuit are first set to sufficiently large bounds, and verification runs followed by failure analysis are repeated. Each verification run performs timed state space enumeration under the given delay bounds, and produces a failure trace if it exists. The failure trace is analyzed, and sufficient timing constraints to prevent the failure is obtained. Then, the delay bounds are tightened according to the timing constraints by using an ILP (Integer Linear Programming) solver. This process terminates when either some delay bounds under which no failure is detected are found or no new delay bounds to prevent the failures can be obtained. The experimental results using a naive implementation show that the proposed method can efficiently handle asynchronous benchmark circuits and nontrivial GasP circuits.
Analog decoding of product codes
A method is presented for analog soft-decision decoding of block product codes (block turbo codes). Extrinsic information is exchanged as analog signals between component row and column decoders. The component MAP decoders use low-power analog computation in subthreshold CMOS circuits to implement the sum-product algorithm. An example decoder design is presented for a (16, 11)/sup 2/ Hamming code.
Cell library for automatic synthesis of analog error control decoders
This paper presents a cell library for automatic synthesis of analog error control decoders. By using some basic cells, analog error control decoders can be automatically synthesized. Also, using automatic synthesis based on this cell library, the circuit performance is not degraded and the circuit is smaller and lower power compared with corresponding canonical designs.
Analysis and Characterization of a Locally-Clocked Module
This thesis describes an evaluation of a locally-clocked module. Locally-clocked modules can be used as synchronous datapath elements in synchronous systems or as asynchronous elements in an asynchronous system. One key element of a locally-clocked module is a stoppable ring oscillator (or stoppable clock). If locally-clocked modules are to be used, their practicality must be quantified. Namely, it must be shown that a reliable and useful stoppable clock can be built. This thesis presents the design and evaluation of a fabricated locally-clocked sequential multiplier. The multiplier is used as a driving example to evaluate local clocks. The design for the stoppable clock is a hybrid of stoppable clocks from previous work. The same gates that make up the critical path of the multiplier are used to make the delay element of the stoppable clock. Although the stoppable clock is meant to track the datapath under a wide range of voltages and temperatures, it is shown that the clock requires tuning to match the critical path sometimes. This is due to the fact that it is difficult to match the critical path exactly. In addition, some temperature and voltage data points cause the cutoff path for the clock to be too slow. This problem is fixed by slowing down the clock. Future designs can focus on speeding up the cutoff path; thus, matching the critical path delay is the only limiting factor on clock frequency. A 20-bit multiplier was fabricated through MOSIS using AMI’s 0.5 μm process. The multiplier consumes 0.468 mm2 and contains 8190 transistors. With a 5 volt power supply, the multiplier runs at 13.3 MHz and consumes 196.6 mW of power, while the stoppable clock runs at 174 MHz. This thesis presents latency and power measurements for the multiplier and stoppable clock in addition to a detailed analysis of stoppable clocks. Process variation is analyzed in that five chips are tested and shown to have little variation in measured values.
Synchronous interlocked pipelines
Locality principles are becoming paramount in controlling advancement of data through pipelined systems. Achieving fine grained power down and progressive pipeline stalls at the local stage level is therefore becoming increasingly, important to enable lower dynamic power consumption while keeping introduced switching noise under control as well as avoiding global distribution of timing critical stall signals. It has long been known that the interlocking properties of as asynchronous pipelined systems have a potential to provide such benefits. However it has not been understood how such interlocking can be achieved in synchronous pipelines. This paper presents a novel technique based on local clock gating and synchronous handshake protocols that achieves stage level interlocking characteristics in synchronous pipelines similar to that of asynchronous pipelines. The presented technique is directly applicable to traditional synchronous pipelines and works equally well for two-phase clocked pipelines based on transparent latches, as well as one-phase clocked pipelines based on master-slave latches.
Direct synthesis of timed circuits from free-choice STGs
Presents a new method to synthesize timed asynchronous circuits directly from the specification without generating a state graph. The synthesis procedure begins with a graph specification with timing constraints. A timing analysis extracts the timed concurrency and timed causality relations directly from the specification. Then, a hazard-free implementation of the specification is synthesized by analyzing precedence graphs which are constructed by using the timed concurrency and timed causality relations. The major result of this work is that the method does not suffer from the state explosion problem, in practice achieves significant reductions in synthesis time for the specifications which have a large state space, and generates synthesized circuits that have nearly the same area as compared to previous timed circuit methods. In particular, this paper shows that a timed circuit-not containing circuit hazards under given timing constraints-can be found by using the relations between signal transitions of the specification. Moreover, the relations can be efficiently found using a heuristic timing analysis algorithm. By allowing significantly larger designs to be synthesized, this work is a step toward the development of high-level synthesis tools for system level asynchronous circuits.
Framework of Timed Trace Theoretic Verification Revisited
This paper develops a framework to support trace theoretic verification of timed circuits and systems. A theoretical foundation for classifying timed traces as either successes or failures is developed. The concept of the semimirror is introduced to allow conformance checking thus supporting hierarchical verification of timed circuits and systems. Finally, we relate our framework to those previously proposed for timing verification.
Modular Synthesis and Verification of Timed Circuits Using Automatic Abstraction
In order to increase performance, circuit designers are beginning to use more aggressive timed circuit designs instead of traditional synchronous static logic designs. Recent design examples have shown that signi cant performance gains are achieved when these aggressive circuit styles are used. Correct operation of these aggressive circuit styles is critically dependent on timing, and in industry they are typically designed by hand. To synthesize and verify timed circuits, the reachable state space of the circuit under the timing constraints needs to be explored. However, complete state space exploration is an exponential problem. State space explosion limits timed circuit designs to small sizes.
Asynchronous Circuit Design
With asynchronous circuit design becoming a powerful tool in the development of new digital systems, circuit designers are expected to have asynchronous design skills and be able to leverage them to reduce power consumption and increase system speed. This book walks readers through all of the different methodologies of asynchronous circuit design, emphasizing practical techniques and real-world applications instead of theoretical simulation. The only guide of its kind, it also features an ftp site complete with support materials. Market: Electrical Engineers, Computer Scientists, Device Designers, and Developers in industry.
Automatic Abstraction for Verification of Timed Circuits and Systems?
This paper presents a new approach for verification of asynchronous circuits by using automatic abstraction. It attacks the state explosion problem by avoiding the generation of a flat state space for the whole design. Instead, it breaks the design into blocks and conducts verification on each of them. Using this approach, the speed of verification improves dramatically.
Analog MAP decoder for (8, 4) Hamming code in subthreshold CMOS
An all-MOS analog implementation of a MAP decoder is presented for the (8, 4) extended Hamming code. This paper describes the design and analysis of a tail-biting trellis decoder implementation using subthreshold CMOS devices. A VLSI test chip has recently returned from fabrication, and preliminary test results indicate accurate decoding up to 20 MBit/s.
Efficient exact two-level hazard-free logic minimization
This paper presents a new approach to two-level hazard free sum-of-products logic minimization. No currently available minimizers for single-output literal-exact two-level hazard-free logic minimization can handle large circuits without synthesis times ranging up over thousands of seconds. The logic minimization approach presented in this paper is based on state graph exploration in conjunction with single-cube cover algorithms. Our algorithm achieves fast logic minimization by using compacted state graphs and cover tables and an efficient algorithm for single-output minimization. Our exact two-level hazard-free logic minimizer finds a minimal number of literal solutions and is significantly faster than existing literal exact methods-over two orders of magnitude faster for the largest extended burst-mode benchmarks to date. This includes a benchmark that has never been possible to solve exactly in a number of literals before.
Analog MAP decoder for (8, 4) Hamming code in subthreshold CMOS
An all-MOS analog implementation of a MAP decoder is presented for the (8, 4) extended Hamming code. This paper describes the design and analysis of a tail-biting trellis decoder implementation using subthreshold CMOS devices. A VLSI test chip has recently returned from fabrication, and preliminary test results indicate accurate decoding up to 20 MBit/s.
A standard-cell self-timed multiplier for energy and area critical synchronous systems
This paper describes the design of a standard-cell self-timed multiplier for use in energy and area critical synchronous systems. The area of this multiplier is bounded by N rather than N/sup 2/ as seen in more traditional combinational parallel array designs, where N is the word size. Energy has a polynomial growth with word size, but has a coefficient that is much smaller than that seen in a combinational array design. Although the multiplier is self-timed, it can be embedded in a synchronous system appearing as a combinational element. This paper presents latency, area, and energy estimates for the multiplier implemented at various word sizes, and compares these numbers with a traditional combinational array multiplier. The self-timed multiplier uses 1/3 the energy and 1/7 the area of the combinational design for a 24-bit word size.
An asynchronous instruction length decoder
This paper describes an investigation of potential advantages and pitfalls of applying an asynchronous design methodology to an advanced microprocessor architecture. A prototype complex instruction set length decoding and steering unit was implemented using self-timed circuits. [The Revolving Asynchronous Pentium/sup (R)/ Processor Instruction Decoder (RAPPID) design implemented the complete Pentium II/sup (R)/ 32-bit MMX instruction set.] The prototype chip was fabricated on a 0.25 /spl mu/m CMOS process and tested successfully. Results show significant advantages - in particular, performance of 2.5-4.5 instructions per nanosecond - with manageable risks using this design technology. The prototype achieves three times the throughput and half the latency, dissipating only half the power and requiring about the same area as the fastest commercial 400 MHz clocked circuit fabricated on the same process.
Timed circuits: a new paradigm for high-speed design
In order to continue to produce circuits of increasing speeds, designers must consider aggressive circuit design styles such as self-resetting or delayed-reset domino circuits used in IBM’s gigahertz processor (GUTS) and asynchronous circuits used in Intel’s RAPPID instruction length decoder. These new timed circuit styles, however, cannot be efficiently and accurately analyzed using traditional static timing analysis methods. This lack of efficient analysis tools is one of the reasons for the lack of mainstream acceptance of these design styles. This paper discusses several industrial timed circuits and gives an overview of our timed circuit design methodology.
Timed circuit verification using TEL structures
Recent design examples have shown that significant performance gains are realized when circuit designers are allowed to make aggressive timing assumptions. Circuit correctness in these aggressive styles is highly timing dependent and, in industry, they are typically designed by hand. In order to automate the process of designing and verifying timed circuits, algorithms for their synthesis and verification are necessary. This paper presents timed event/level (℡) structures, a specification formalism for timed circuits that corresponds directly to gate-level circuits. It also presents an algorithm based on partially ordered sets to make the state-space exploration of ℡ structures more tractable. The combination of the new specification method and algorithm significantly improves efficiency for gate-level timing verification. Results on a number of circuits, including many from the recently published gigahertz unit Test Site (guTS) processor from IBM indicate that modules of significant size can be verified using a level of abstraction that preserves the interesting timing properties of the circuit. Accurate circuit level verification allows the designer to include less margin in the design, which can lead to increased performance.
Achieving fast and exact hazard-free logic minimization of extended burst-mode gC finite state machines
This paper presents a new approach to two-level hazard-free logic minimization in the context of extended burst-mode finite state machine synthesis targeting generalized C-elements (gC). No currently available minimizers for literal-exact two-level hazard-free logic minimization of extended burst-mode gC controllers can handle large circuits without synthesis times ranging up over thousands of seconds. Even existing heuristic approaches take too much time when iterative exploration over a large design space is required and do not yield minimum results. The logic minimization approach presented in this paper is based on state graph exploration in conjunction with single-cube cover algorithms, an approach that has not been considered for minimization of extended burst-mode finite state machines previously. Our algorithm achieves very fast logic minimization by introducing compacted state graphs and cover tables and an efficient single-cube cover algorithm for single-output minimization. Our exact logic minimizer finds minimal number of literal solutions to all currently available benchmarks, in less than one second on a 333 MHz microprocessor-more than three orders of magnitude faster than existing literal exact methods, and over an order of magnitude faster than existing heuristic methods for the largest benchmarks. This includes a benchmark that has never been possible to solve exactly in number of literals before.
Interfacing synchronous and asynchronous modules within a high-speed pipeline
This paper describes a new technique for integrating asynchronous modules within a high-speed synchronous pipeline. Our design eliminates potential metastability problems by using a clock generated by a stoppable ring oscillator, which is capable of driving the large clock load found in present day microprocessors. Using the ATACS design tool, we designed highly optimized transistor-level circuits to control the ring oscillator and generate the clock and handshake signals with minimal overhead. Our interface architecture requires no redesign of the synchronous circuitry. Incorporating asynchronous modules in a high-speed pipeline improves performance by exploiting data-dependent delay variations. Since the speed of the synchronous circuitry tracks the speed of the ring oscillator under different processes, temperatures, and voltages, the entire chip operates at the speed dictated by the current operating conditions, rather than being governed by the worst case conditions. These two factors together can lead to a significant improvement in average-case performance. The interface design is simulated using the 0.6-/spl mu/m HP CMOS14B process in HSPICE.
Interfacing synchronous and asynchronous modules within a high-speed pipeline
This paper describes a new technique for integrating asynchronous modules within a high-speed synchronous pipeline. Our design eliminates potential metastability problems by using a clock generated by a stoppable ring oscillator, which is capable of driving the large clock load found in present day microprocessors. Using the ATACS design tool, we designed highly optimized transistor-level circuits to control the ring oscillator and generate the clock and handshake signals with minimal overhead. Our interface architecture requires no redesign of the synchronous circuitry. Incorporating asynchronous modules in a high-speed pipeline improves performance by exploiting data-dependent delay variations. Since the speed of the synchronous circuitry tracks the speed of the ring oscillator under different processes, temperatures, and voltages, the entire chip operates at the speed dictated by the current operating conditions, rather than being governed by the worst case conditions. These two factors together can lead to a significant improvement in average-case performance. The interface design is simulated using the 0.6-/spl mu/m HP CMOS14B process in HSPICE.
Timed state space exploration using POSETs
This paper presents a new timing analysis algorithm for efficient state space exploration during the synthesis of timed circuits or the verification of timed systems. The source of the computational complexity in the synthesis or verification of a timed system is in finding the reachable timed state space. We introduce a new algorithm which utilizes geometric regions to represent the timed state space and partially ordered sets (POSET’s) to minimize the number of regions necessary. This algorithm operates on specifications sufficiently general to describe practical circuits, as well as other timed systems. The algorithm is applied to several examples showing significant improvement in runtime and memory usage.
Stochastic cycle period analysis in timed circuits
This paper presents a technique to estimate the stochastic cycle period (SCP), a performance metric for timed asynchronous circuits. This technique uses timed stochastic Petri nets (TSPN) which support choice and arbitrary delay distributions. The SCP is the delay of the average path in a TSPN when represented as a sum of weighted place delays. A place delay is the expected value of its associated distribution and its weight denotes its importance in the average path of the TSPN. The approach analyzes finite execution traces of the TSPN to derive an expression for the weight values in the SCP. The weights can be analyzed with basic statistics to within an arbitrary error bound. This paper demonstrates the use of the SCP to aggressively optimize timed asynchronous circuits for improved average-case performance by reducing transistor counts, reordering input pins at gates, and skewing transistor sizes to favor important transitions. Each optimization effort is directed to improve the average-case delay in the circuit at the possible expense of the worst-case delay.
Apparatus and Method for Parallel Processing and Self-Timed Serial Marking of Variable Length Instructions
Optimal parallelization of necessarily serial operations is performed by speculative parallel processing and propagation of serial marking signals to indicate valid data. An exemplary instruction marking circuit for a computer system implementing such optimization includes a series of columns, each column corresponding ton one byte of a fixed length instruction line, and a length decoder in each column. Each length decoder receives a byte of the respective column, and performs a length decode independently of the other length decoder. The length decoder asserts a length signal indicative of an instruction length when the byte is the first byte of an instruction. A marking unit arrangement is coupled to the length decoders, and operates to mark each column containing a first byte of an instruction as a function of the length signals asserted by the length decoders.
Direct synthesis of timed asynchronous circuits
This paper presents a new method to synthesize timed asynchronous circuits directly from the specification without generating a state graph. The synthesis procedure begins with a deterministic graph specification with timing constraints. A timing analysis extracts the timed concurrency and timed causality relations between any two signal transitions. Then, a hazard-free implementation of the specification is synthesized by analyzing precedence graphs which are constructed by using the timed concurrency and timed causality relations. The major result of this work is that the method does not suffer from the state explosion problem, achieves significant reductions in synthesis time, and generates synthesized circuits that have nearly the same area as compared to previous timed circuit methods. In particular, this paper shows that a timed circuit-not containing circuit hazards under given timing constraints-can be found by using the relations between signal transitions of the specification. Moreover, the relations can be efficiently found using a heuristic timing analysis algorithm. By allowing significantly larger designs to be synthesized, this work is a step towards the development of high-level synthesis tools for system level asynchronous circuits.
Architectural synthesis of timed asynchronous systems
Describes a new method for the architectural synthesis of timed asynchronous systems. Due to the variable delays associated with asynchronous resources, implicit schedules are created by the addition of supplementary constraints between resources. Since the number of schedules grows exponentially with respect to the size of the given data flow graph, pruning techniques are introduced which dramatically improve the run-time without significantly affecting the quality of the results. Using a combination of data and resource constraints, as well as an analysis of bounded delay information, our method determines the minimum number of resources and registers needed to implement a given schedule. Results are demonstrated using some high-level synthesis benchmark circuits and an industrial example.
Apparatus and method for self-timed marking of variable length instructions having length-affecting prefix bytes
A self-timed instruction marking circuit includes a prefix handling system for processing instruction bytes having prefix bytes. Length decoders receive instruction data bytes, and perform length decoding independently of the other length decoders in the instruction marking circuit. A length decoder determines whether a byte being processed is a prefix byte to an instruction. If a length-affecting prefix byte is found, the length decoder signals a subsequent length decoder to indicate that a prefix byte has been found. The subsequent length decoder uses the prefix signal to appropriately length decode the byte being processed by the subsequent length decoder. Signals are provided to continue the self-timed marking process. Prefix handling may also be used in a multiple marking unit configuration of an instruction marking circuit.
Algorithms for Synthesis and Verification of Timed Circuits and Systems
In order to increase performance, circuit designers are beginning to move away from traditional, synchronous designs based on static logic. Recent design examples have shown that significant performance gains are realized when aggressive circuit styles are used. Circuit correctness in these aggressive circuit styles is highly timing dependent, and in industry they are typically designed by hand. In order to automate the process of designing and verifying timed circuits, algorithms to explore the reachable state space of the circuit under the timing constraints are necessary. This thesis presents a new specification method for timed circuits, timed event/level (TEL) structures, and new algorithms for exploring a timed state space. The TEL structure specification allows the designer to specify behavior controlled by signal transitions, which is best for representing sequencing, and behavior controlled by signal levels, which is best for representing gate level circuits. This thesis also presents algorithms based on partially ordered sets (POSETs) that explores the timed state space of the TEL structure. Results using the new specification method and algorithms show orders of magnitude improvement over previous techniques in both speed and memory performance. The algorithms have also been successfully applied to several circuit examples from the recently published experimental Gigahertz processor developed at IBM. The speed and memory performance improvements of the algorithm allow automatic synthesis and verification of complex timed circuits, making them an attractive design alternative.
Efficient self-timed marking of lengthy variable length instructions
A self-timed instruction marking circuit includes a long instruction processing system to divide long instruction processing between two columns of the instruction marking circuit. Length decoders are interconnected across columns to signal the presence and length of long instructions. Self-timed marking can continue without alteration. The number of connections required by the instruction marking circuit are reduced. The marking process can be optimized to efficiently process all instructions by setting the definition of a long instruction such that commonly executed instructions are not included.
Branch instruction handling in a self-timed marking system
An instruction execution pipeline in a computer system having variable-length instructions uses branch prediction to perform self-timed marking of instructions prior to decoding. Branch handling logic is provided in an instruction marking circuit to directly mark a target instruction of a predicted branch as the next instruction to be decoded. Additionally, a branch target FIFO may be used to store information about the location of the target instruction in the instruction stream.
Stochastic cycle period analysis in timed circuits
This paper presents a technique to estimate the stochastic cycle period (SCP), a performance metric for timed asynchronous circuits. This technique uses timed stochastic Petri nets (TSPN) which support choice and arbitrary delay distributions. The SCP is the delay of the average path in a TSPN when represented as a sum of weighted place delays. A place delay is the expected value of its associated distribution and its weight denotes its importance in the average path of the TSPN. The approach analyzes finite execution traces of the TSPN to derive an expression for the weight values in the SCP. The weights can be analyzed with basic statistics to within an arbitrary error bound. This paper demonstrates the use of the SCP to aggressively optimize timed asynchronous circuits for improved average-case performance by reducing transistor counts, reordering input pins at gates, and skewing transistor sizes to favor important transitions. Each optimization effort is directed to improve the average-case delay in the circuit at the possible expense of the worst-case delay.
Direct synthesis of timed asynchronous circuits
This paper presents a new method to synthesize timed asynchronous circuits directly from the specification without generating a state graph. The synthesis procedure begins with a deterministic graph specification with timing constraints. A timing analysis extracts the timed concurrency and timed causality relations between any two signal transitions. Then, a hazard-free implementation of the specification is synthesized by analyzing precedence graphs which are constructed by using the timed concurrency and timed causality relations. The major result of this work is that the method does not suffer from the state explosion problem, achieves significant reductions in synthesis time, and generates synthesized circuits that have nearly the same area as compared to previous timed circuit methods. In particular, this paper shows that a timed circuit-not containing circuit hazards under given timing constraints-can be found by using the relations between signal transitions of the specification. Moreover, the relations can be efficiently found using a heuristic timing analysis algorithm. By allowing significantly larger designs to be synthesized, this work is a step towards the development of high-level synthesis tools for system level asynchronous circuits.
POSET timing and its application to the synthesis and verification of gate-level timed circuits
This paper presents a new algorithm for timed state-space exploration, POSET timing, POSET timing improves upon geometric methods by utilizing concurrency and causality information to dramatically reduce the number of geometric regions needed to represent the timed state space. The utility of POSET timing is illustrated by its application to the automatic synthesis and verification of gate-level timed circuits. Timed circuits are a class of asynchronous circuits that incorporate explicit timing information in the specification which is used throughout the synthesis procedure to optimize the design. Using POSET timing, our synthesis procedure derives a timed circuit that is hazard-free. The circuit uses only basic gates to facilitate the mapping to semi-custom components, such as standard-cells and gate-arrays. The resulting gate-level timed circuit implementations are 30%-40% smaller and 30%-50% faster than those produced using other asynchronous design methodologies. This paper also demonstrates that timed designs can be smaller and faster than their synchronous counterparts. The POSET timing algorithm cannot only efficiently verify our synthesized circuits but also a wide collection of large, highly concurrent timed circuits and systems that could not previously be verified using traditional techniques.
Stochastic Cycle Period Analysis in Timed Circuits
This thesis presents a method of deriving a performance metric for timed asynchronous circuits called a stochastic cycle period, which uses analytical techniques combined with simulation to capture the stochastic profile of the system. The stochastic cycle period is constructed by finding transition and steady-state probabilities in a reachability graph of the timed circuit. The transition and steady-state probabilities are used to obtain trigger probabilities in the circuit implementation. The trigger probabilities are employed in a timing simulation to construct the stochastic cycle period of the timed specification. Since this performance metric is a stochastic profile of the circuit behavior with regards to its individual components, synthesis optimization efforts can be focused on areas that significantly improve the expected cost of a cycle in the system. This thesis presents some case studies where the metric is used to evaluate and improve designs. The studies show the potentia…
Verification of delayed-reset domino circuits using ATACS
This paper discusses the application of the timing analysis tool ATACS to the high performance, self-resetting and delayed-reset domino circuits being designed at IBM’s Austin Research Laboratory. The tool, which was originally developed to deal with asynchronous circuits, is well suited to the self-resetting style since internally, a block of self-resetting or delayed-reset domino logic is asynchronous. The circuits are represented using timed event/level structures. These structures correspond very directly to gate level circuits, making the translation from a transistor schematic to a ℡ structure straightforward. The state-space explosion problem is mitigated using an algorithm based on partially ordered sets (POSETs). Results on a number of circuits from the recently published guTS (gigahertz unit Test Site) processor from IBM indicate that modules of significant size can be verified with ATACS using a level of abstraction that preserves the interesting timing properties of the circuit. Accurate circuit level verification allows the designer to include less margin in the design, which can lead to increased performance.
RAPPID: an asynchronous instruction length decoder
This paper describes an investigation of potential advantages and risks of applying an aggressive asynchronous design methodology to Intel Architecture. RAPPID ("Revolving Asynchronous Pentium(R) Processor Instruction Decoder"), a prototype IA32 instruction length decoding and steering unit, was implemented using self-timed techniques. RAPPID chip was fabricated on a 0.25 /spl mu/ CMOS process and tested successfully. Results show significant advantages-in particular, performance of 2.5-4.5 instructions/nS-with manageable risks using this design technology. RAPPID achieves three times the throughput and half the latency, dissipating only half the power and requiring about the same area as an existing 400 MHz clocked circuit.
Timed circuit synthesis using implicit methods
The design and synthesis of asynchronous circuits is gaining importance in both the industrial and academic worlds. Timed circuits are a class of asynchronous circuits that incorporate explicit timing information in the specification. This information is used throughout the synthesis procedure to optimize the design. In order to synthesize a timed circuit, it is necessary to explore the timed state space of the specification. The memory required to store the timed state space of a complex specification can be prohibitive for large designs when explicit representation methods are used. This paper describes the application of BDDs and MTBDDs to the representation of timed state spaces and the synthesis of timed circuits. These implicit techniques significantly improve the memory efficiency of timed state space exploration and allow more complex designs to be synthesized. Implicit methods also allow the derivation of solution spaces containing all valid solutions to the synthesis problem facilitating subsequent optimization and technology mapping steps.
Architectural-Level Synthesis of Asynchronous Systems
Asynchronous circuit design has the potential to produce circuits superior to those of synchronous circuit design. Current synchronous methods of architectural-level synthesis do not exploit properties inherent to asynchronous circuits. This research describes potential optimizations and techniques that can be applied to the architectural-level design of asynchronous systems. The proposed methods take advantage of asynchronous circuit properties such as data-dependent delays, modularity, and composiblity. The optimization problems of scheduling and allocation are studied. For scheduling, some counterintuitive properties of delays in a system are shown. The design space is studied and several filters to reduce the size of the design space are proposed. To evaluate and test these ideas the CAD tool Mercury was developed and is described in detail. Mercury is unique in that it can take an abstract model of a design, in this case a data ow graph, and from that generate both an optimal structural view of an asynchronous datapath for the design, as well as the necessary behavioral control to operate that datapath. Several case studies are presented utilizing the tool and methods to illustrate the practical aspects of this work.
Verification of timed systems using POSETs
This paper presents a new algorithm for efficiently verifying timed systems. The new algorithm represents timing information using geometric regions and explores the timed state space by considering partially ordered sets of events rather than linear sequences. This approach avoids the explosion of timed states typical of highly concurrent systems by dramatically reducing the ratio of timed states to untimed states in a system. A general class of timed systems which include both event and level causality can be specified and verified. This algorithm is applied to several recent timed benchmarks showing orders of magnitude improvement in runtime and memory usage.
Specification and Compilation of Timed Systems
This thesis presents a framework for the specification and compilation of modules in a system that uses different synchronization paradigms. These timed systems are described by using timed handshaking expansions (HSE) and a standard hardware description language, namely VHDL. Synthesizable subsets of these languages are defined to include constructs for describing timing behaviors, as well as, sequencing, concurrency, choice and looping. A new formal semantic model, timed event/level structures, is used to define the behaviors specified by the synthesizable subsets. A compiler is developed to translate the HSE and VHDL specifications to timed event/level structures. This compiler is integrated into ATACS, a synthesis tool for timed circuits. Finally we demonstrate our methodology on a practical example, an asynchronous implementation of the Maxlist algorithm.
Implicit Methods for Timed Circuit Synthesis
The design and synthesis of asynchronous circuits is gaining importance in both the industrial and academic worlds. Timed circuits are a class of asynchronous circuits that incorporate explicit timing information in the specification. This information is used throughout the synthesis procedure to optimize the design. In order to synthesize a timed circuit, it is necessary to explore the timed state space of the specification. The memory required to store the timed state space of a complex specification can be prohibitive for large designs when explicit representation methods are used. This thesis describes the application of BDDs and MTBDDs to the representation of timed state spaces and the synthesis of timed circuits. These implicit techniques significantly improve the memory eficiency of timed state space exploration and allow more complex designs to be synthesized.
Covering conditions and algorithms for the synthesis of speed-independent circuits
This paper presents theory and algorithms for the synthesis of standard C-implementations of speed-independent circuits. These implementations are block-level circuits which may consist of atomic gates to perform complex functions in order to ensure hazard freedom. First, we present Boolean covering conditions that guarantee that the standard C-implementations operate correctly. Then, we present two algorithms that produce optimal solutions to the covering problem. The first algorithm is always applicable, but does not complete on large circuits. The second algorithm, motivated by our observation that our covering problem can often be solved with a single cube, finds the optimal single-cube solution when such a solution exists. When applicable, the second algorithm is dramatically more efficient than the first, more general algorithm. We present results for benchmark specifications which indicate that our single-cube algorithm is applicable on most benchmark circuits and reduces run times by over an order of magnitude. The block-level circuits generated by our algorithms are a good starting point for tools that perform technology mapping to obtain gate-level speed-independent circuits.
Average-case optimized technology mapping of one-hot domino circuits
This paper presents a technology mapping technique for optimizing the average-case delay of asynchronous combinational circuits implemented using domino logic and one-hot encoded outputs. The technique minimizes the critical path for common input patterns at the possible expense of making less common critical paths longer. To demonstrate the application of this technique, we present a case study of a combinational length decoding block, an integral component of an Asynchronous Instruction Length Decoder (AILD) which can be used in Pentium(R) processors. The experimental results demonstrate that the average-case delay of our mapped circuits can be dramatically lower than the worst-case delay of the circuits obtained using conventional worst-case mapping techniques.
Efficient timing analysis algorithms for timed state space exploration
This paper presents new timing analysis algorithms for efficient state space exploration during timed circuit synthesis. Timed circuits are a class of asynchronous circuits that incorporate explicit timing information in the specification which is used throughout the synthesis procedure to optimize the design. Much of the computational complexity in the synthesis of timed circuits currently is in finding the reachable timed state space. We introduce new algorithms which utilize geometric regions to represent the timed state space and partial orders to minimize the number of regions necessary. These algorithms operate on specifications sufficiently general to describe practical circuits.
An asynchronous implementation of the maxlist algorithm
We present an efficient asynchronous VLSI architecture for calculating running maximum or minimum values over a sliding window. Running maximums or minimums are very useful for many signal and image processing tasks. Our architecture performs the calculation using the MAXLIST algorithm. In order to take advantage of the wide delay variations due to data-dependencies and operating conditions, an asynchronous approach is taken to achieve higher performance and lower power. Simulation results demonstrate that our asynchronous architecture is significantly faster than existing and potential synchronous architectures.
Computer Aided Synthesis and Verification of Gate-Level Timed Circuits
In recent years, there has been a resurgence of interest in the design of asynchronous circuits due to their ability to eliminate clock skew problems, achieve average case performance, adapt to processing and environmental variations, provide component modularity, and lower system power requirements. Traditional academic asynchronous designs methods use unbounded delay assumptions, resulting in circuits that are verifiable, but ignore timing for simplicity, leading to unnecessarily conservative designs. In industry, however, timing is critical to reduce both chip area and circuit delay. Due to a lack of formal methods that handle timing information correctly, circuits with timing constraints usually require extensive simulation to gain confidence in the design. This thesis bridges this gap by introducing timed circuits in which explicit timing information is incorporated into the specification and utilized throughout the design procedure to optimize the implementation. Our timed circu…
Technology mapping of timed circuits
This paper presents an automated procedure for the technology mapping of timed circuits to practical gate libraries. Timed circuits are a class of asynchronous circuits that incorporate explicit timing information in the specification which is used throughout the design process to optimize the implementation. Our procedure begins with a timed specification and a delay-annotated gate library description which must include 2-input AND gates, OR gates, and C-elements, but optionally can include higher-fanin gates, AND-OR-INVERT blocks, and generalized C-elements. Our procedure first generates a technology-independent timed circuit netlist composed of possibly high-fanin AND gates, OR gates, and 2-input C-elements. The procedure then investigates simultaneous decompositions of all high-fanin gates by adding state variables to the the specification and performing resynthesis. Although multiple decompositions are explored timing information is utilized to significantly reduce their number. Once all gates are sufficiently decomposed, the netlist can be mapped to the given gate library, taking advantage of any compact complex gates available. The decomposition and resynthesis steps have been fully automated within the synthesis tool ATACS and we present results for several examples.
Automatic synthesis of gate-level timed circuits with choice
This paper presents a CAD tool for the automatic synthesis of gate-level timed circuits from general specifications to basic gates such as AND gates, OR gates, and C-elements. Timed circuits are a class of asynchronous circuits that incorporate explicit timing information in the specification which is used throughout the synthesis procedure to optimize the design. Our procedure begins with a textual specification capable of specifying conditional operation, or choice. This specification is systematically transformed to a graphical representation which can be analyzed using an exact and efficient timing analysis algorithm to find the reachable stale space. From this state space, a timed circuit that is hazard-free at the gate-level is derived, facilitating the use of semi-custom components, such as standard-cells and gate-arrays. Because timing information is used to guide the synthesis to reduce circuit complexity while guaranteeing correct operation, the resulting timed circuit implementations are up to 40 percent smaller and 50 percent faster than those produced using other design methodologies.
Automatic verification of timed circuits
This paper presents a new formalism and a new algorithm for verifying timed circuits. The formalism, called orbital nets, allows hierarchical verification based on a behavioral semantics of timed trace theory. We present improvements to a geometric timing algorithm that take advantage of concurrency by using partial orders to reduce the time and space requirements of verification. This algorithm has been fully automated and incorporated into a design system for timed circuits, and experimental results demonstrate that this verification algorithm is practical for realistic examples.
Synthesis of timed asynchronous circuits
The authors present a systematic procedure for synthesizing timed asynchronous circuits using timing constraints dictated by system integration, thereby facilitating natural interaction between synchronous and asynchronous circuits. Their timed circuits also tend to be more efficient, in both speed and area, compared with traditional asynchronous circuits. The synthesis procedure begins with a cyclic graph specification to which timing constraints can be added. First, the cyclic graph is unfolded into an infinite acyclic graph. Then, an analysis of two finite subgraphs of the infinite acyclic graph detects and removes redundancy in the original specification based on the given timing constraints. From this reduced specification, an implementation that is guaranteed to function correctly under the timing constraints is systematically synthesized. With practical circuit examples, it is demonstrated that the resulting timed implementation is significantly reduced in complexity compared with implementations previously derived using other methodologies.textlesstextgreater
Synthesis of timed asynchronous circuits
A synthesis method that utilizes timing constraints to generate timed asynchronous circuits is presented. By unfolding the cyclic graph specification of an asynchronous circuit into an infinite acyclic graph, it is possible to use efficient algorithms to analyze the given timing constraints. A sufficient condition for the removal of redundancy in the specification is derived. Because of this condition, it is only necessary to analyze a finite subgraph of the infinite acyclic graph for derivation of a correct implementation. A systematic synthesis procedure that further optimizes the implementation based on the timing constraints is applied to the reduced specification. It is shown that the resulting timed implementation can be significantly reduced in complexity from its speed-independent counterpart while remaining hazard-free under the given timing constraints.textlesstextgreater