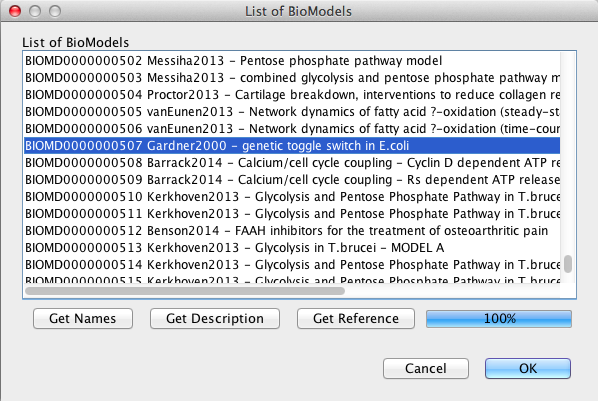

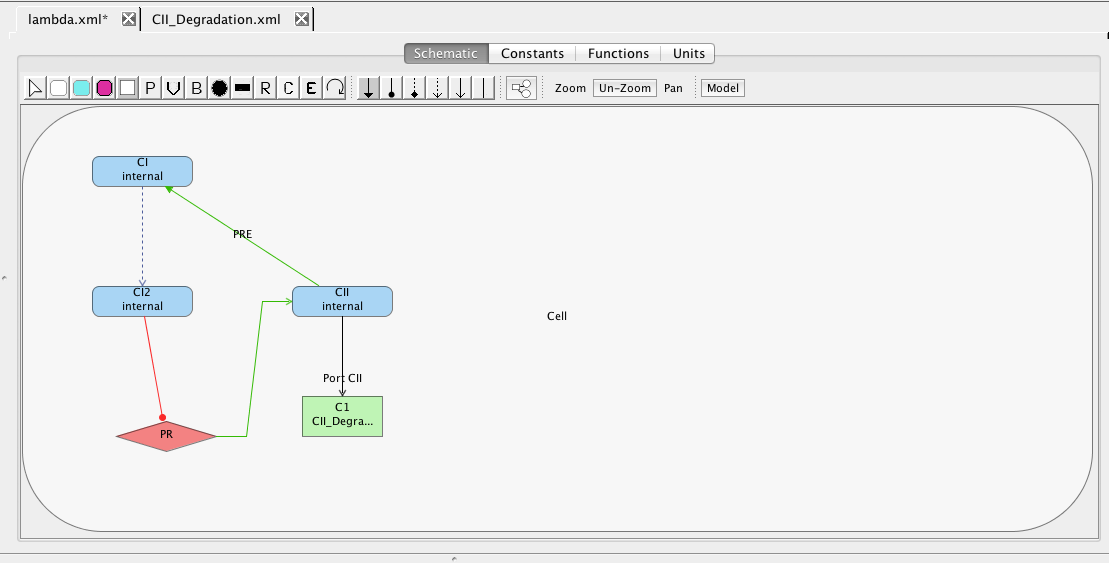
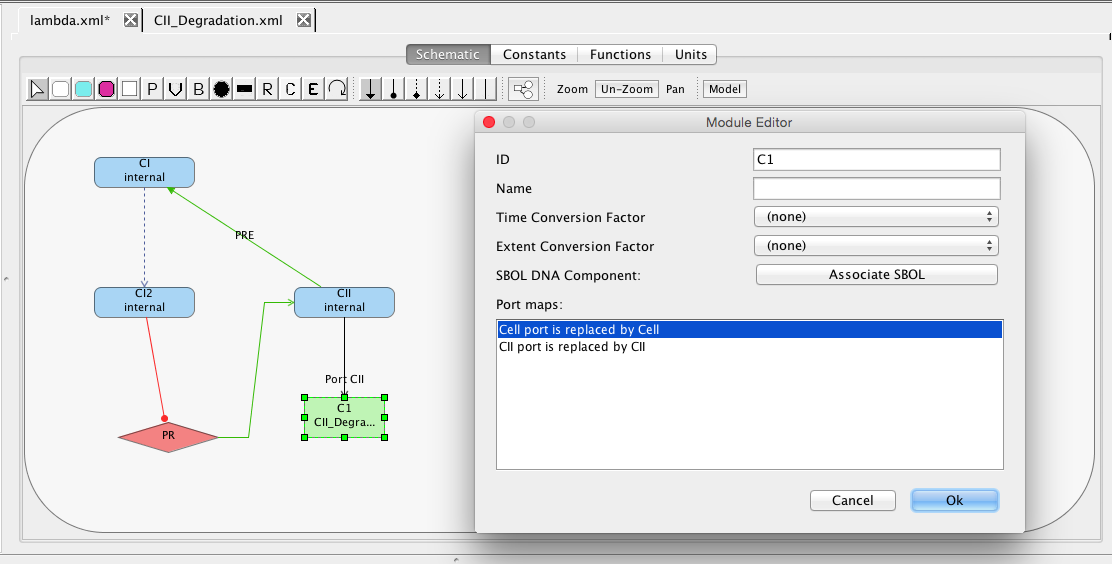
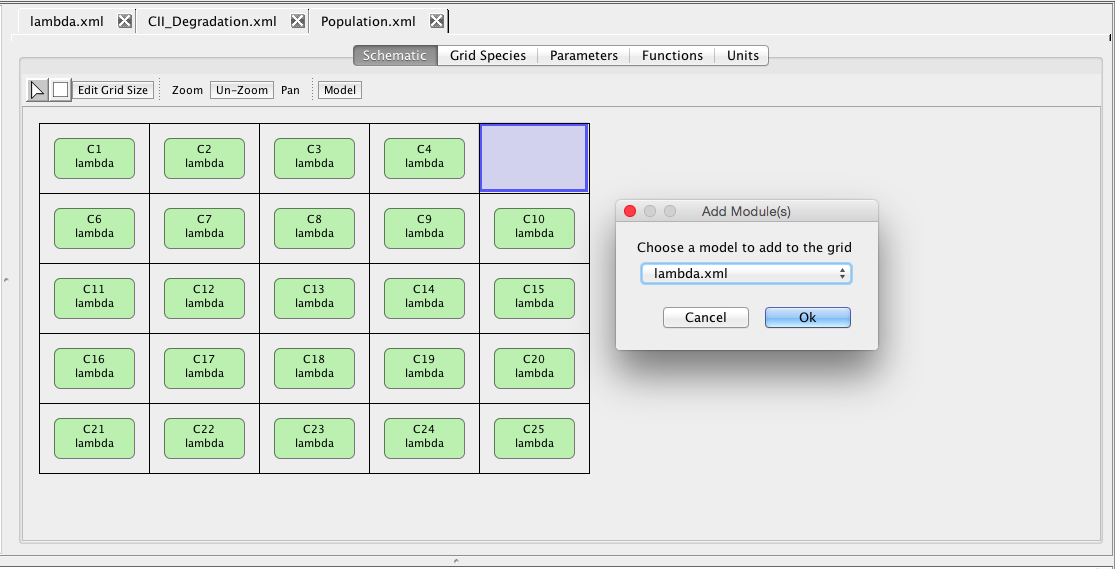
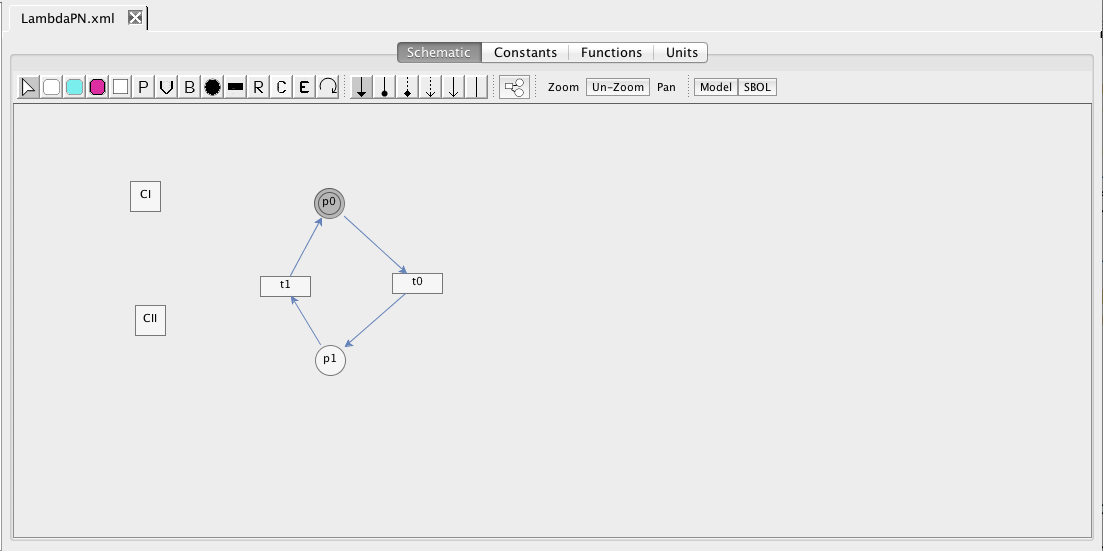
| Algebraic | left-hand side is zero | 0 = f(W) |
| Assignment | left-hand side is a scalar | x = f(W) |
| Rate | left-hand side is a rate-of-change | [dx/dt] = f(W) |
| ID | Default Value | Units | Structure | Description |
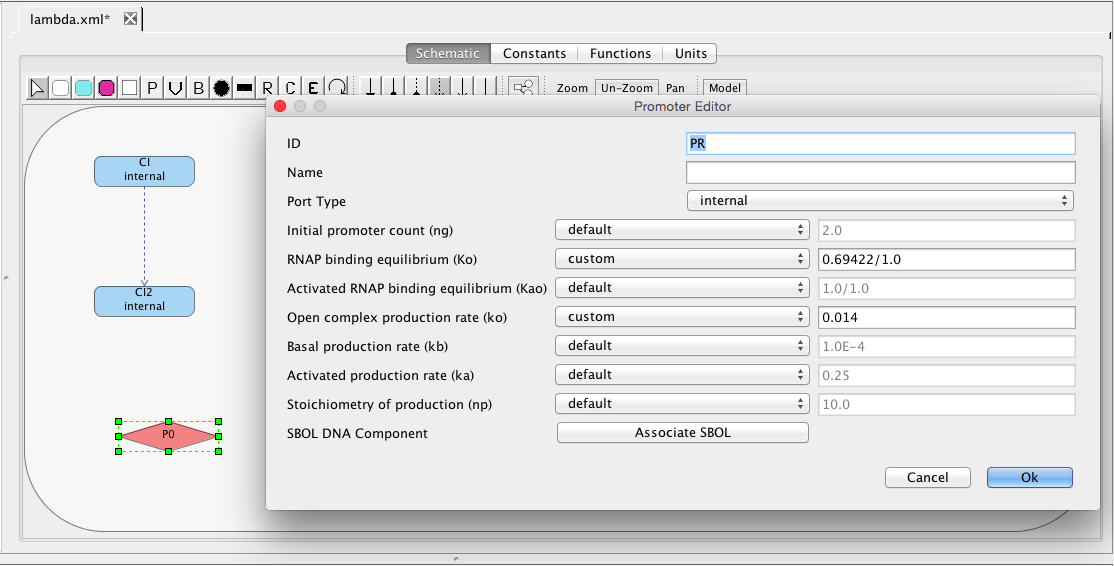
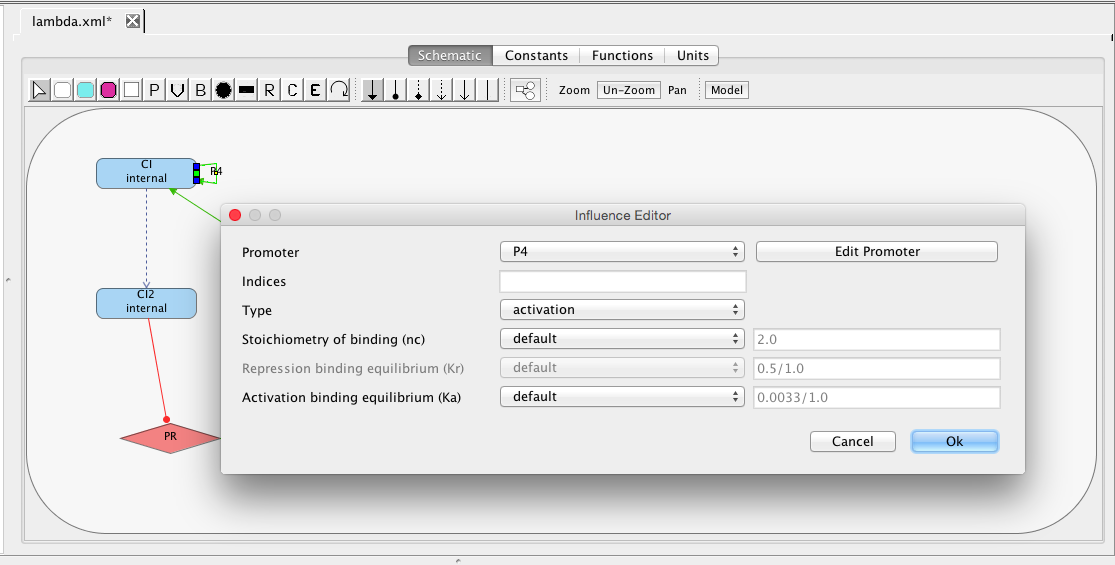
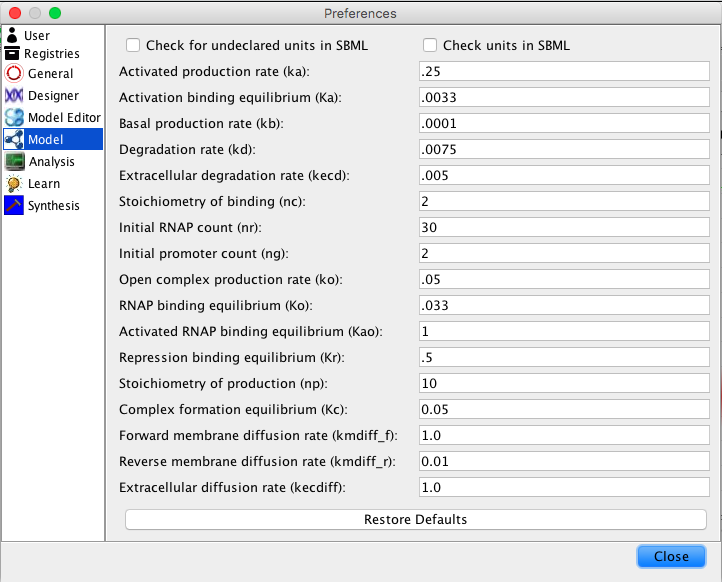
| ka | 0.25 | [1/(sec)] | promoter | Activated production rate |
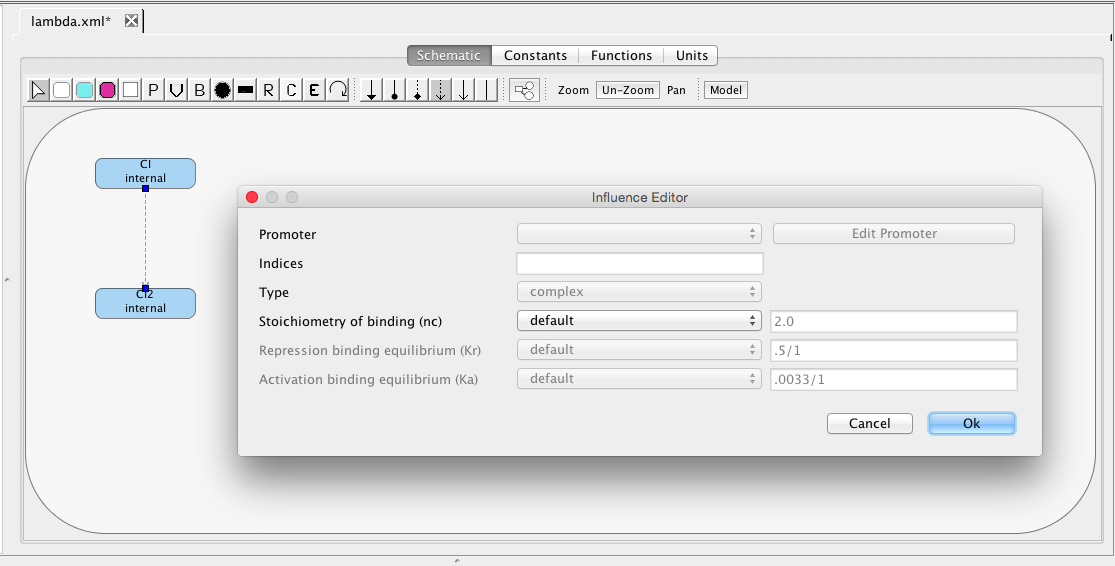
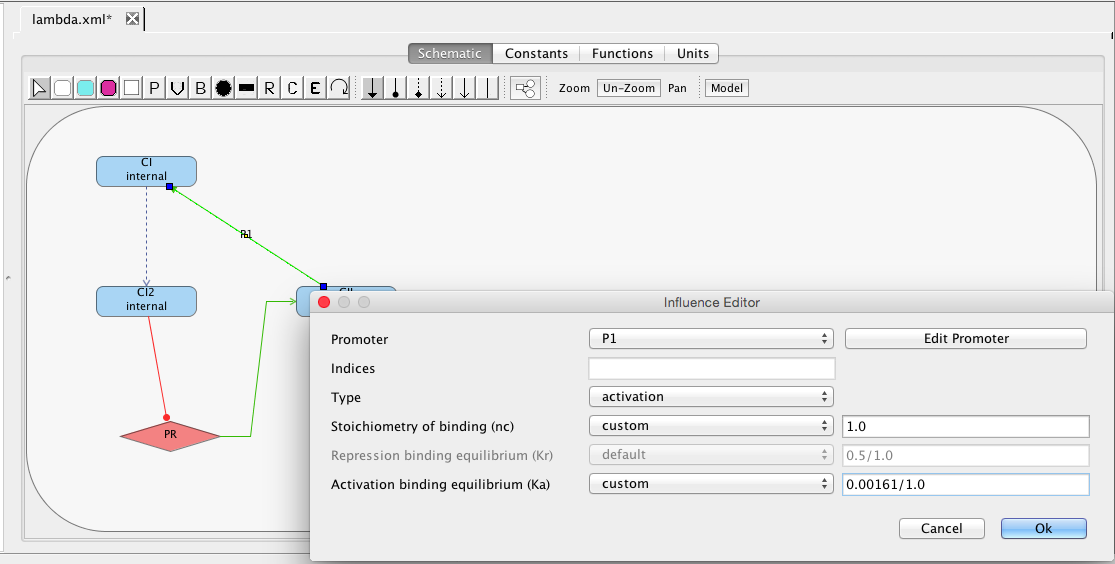
| Ka | 0.0033 | [1/(molecule(nc+1))] | influence | Activation binding equilibrium |
| kb | 0.0001 | [1/(sec)] | promoter | Basal production rate |
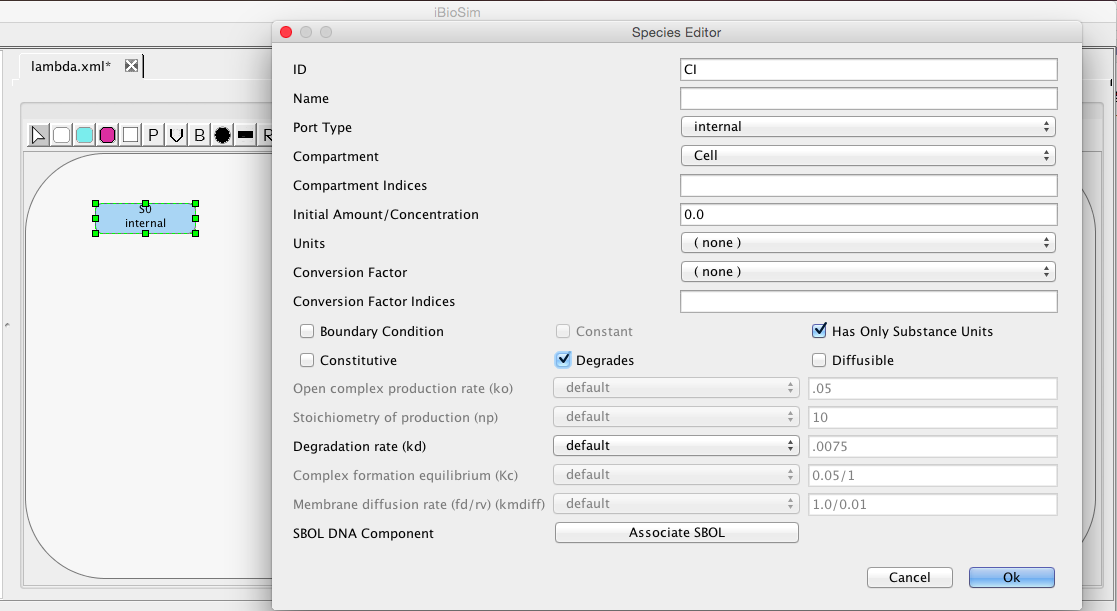
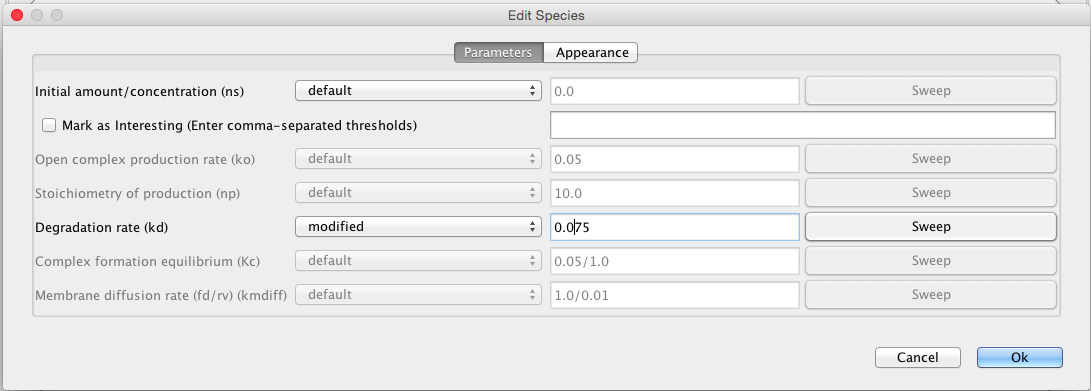
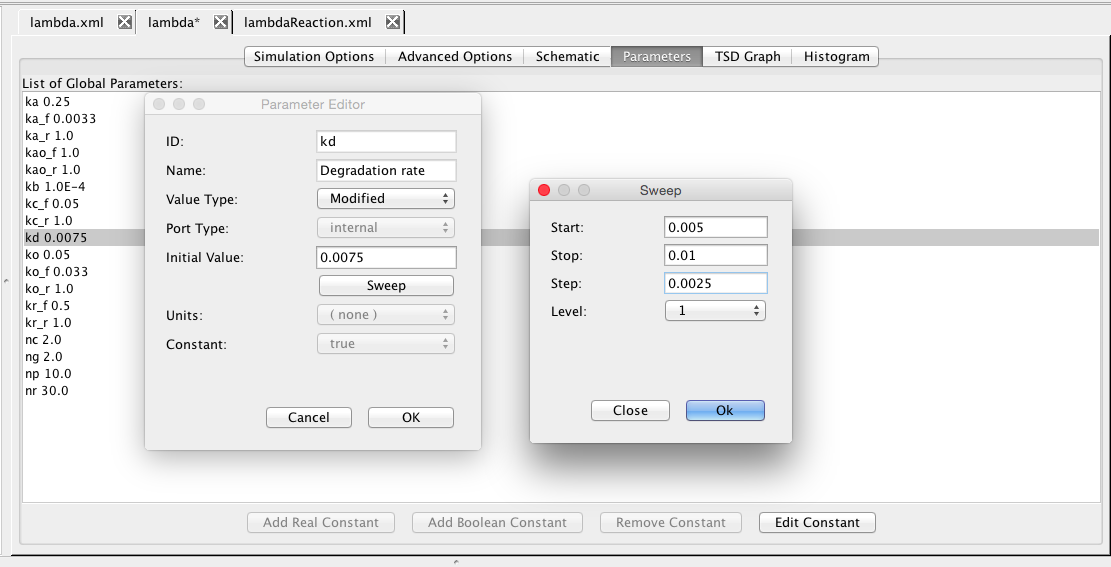
| kd | 0.0075 | [1/(sec)] | species | Degradation rate |
| kecd | 0.005 | [1/(sec)] | species | Extracellular degradation rate |
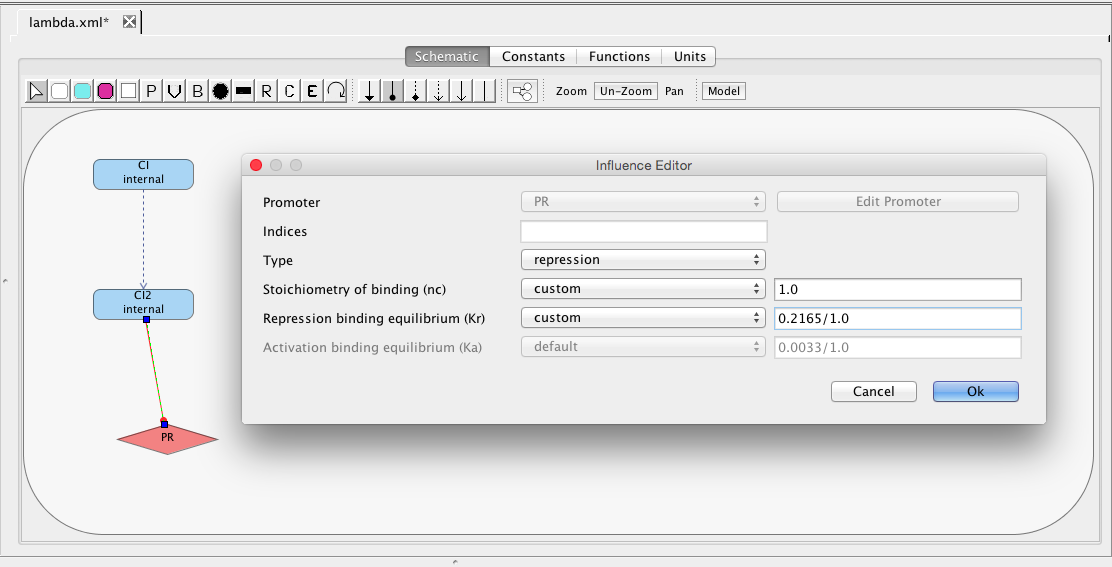
| nc | 2 | molecule | influence | Stoichiometry of binding |
| nr | 30 | molecule | model | Initial RNAP count |
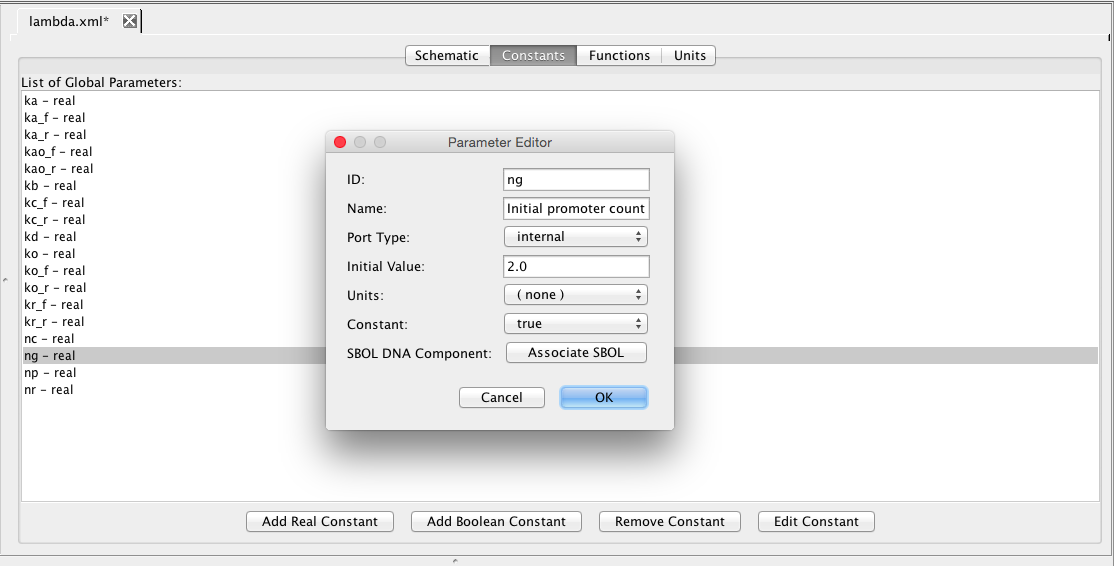
| ng | 2 | molecule | promoter | Initial promoter count |
| ko | 0.05 | [1/(sec)] | promoter | Open complex production rate |
| Ko | 0.033 | [1/(molecule)] | promoter | RNAP binding equilibrium |
| Kao | 1 | [1/(molecule)] | promoter | Activated RNAP binding equilibrium |
| Kr | 0.5 | [1/(moleculenc)] | influence | Repression binding equilibrium |
| np | 10 | molecule | promoter | Stoichiometry of production |
| Kc | 0.05 | [1/(molecule)] | species | Complex formation equilibrium |
| kmdiff_f | 1.0 | [1/(molecule)] | species | Forward membrane diffusion rate |
| kmdiff_r | 0.01 | [1/(molecule)] | species | Reverse membrane diffusion rate |
| kecdiff | 1.0 | [1/(molecule)] | species | Extracellular diffusion rate |
|
| ampere | farad | joule | lux | radian | volt |
| avogadro | gram | katal | metre | second | watt |
| bacquerel | gray | kelvin | mole | siemens | weber |
| candela | henry | kilogram | newton | sievert | |
| coulomb | hertz | litre | ohm | steradian | |
| dimensionless | item | lumen | pascal | tesla |
| Type | Method ID | Description |
| ODE | Euler | The forward Euler Method |
| ODE | gear1 | Gear Method M=1 |
| ODE | gear2 | Gear Method M=2 |
| ODE | rk4imp | Implicit 4th order Runge-Kutta at Gaussian points |
| ODE | rk8pd | Embedded Runge-Kutta Prince-Dormand (8,9) method |
| ODE | rkf45 | Embedded Runge-Kutta-Fehlberg (4, 5) method |
| ODE | Runge-Kutta-Fehlberg | Runge-Kutta-Fehlberg method (java) |
| Monte Carlo | gillespie | SSA-direct method |
| Monte Carlo | SSA-Hierarchical | SSA-direct method on hierarchical models (java) |
| Monte Carlo | SSA-Direct | SSA-direct method (java) |
| Monte Carlo | SSA-CR | SSA composition and rejection method (java) |
| Monte Carlo | iSSA | incremental SSA |
| Monte Carlo | interactive | Interactive SSA-direct method |
| Monte Carlo | emc-sim | Uses jump count as next reaction time |
| Monte Carlo | bunker | Uses mean for next reaction time |
| Monte Carlo | nmc | Uses normally distributed next reaction time |
| Markov | steady-state | Steady-state Markov chain analysis |
| Markov | transient | Transient Markov chain analysis |
| Markov | reachability | Only perform reachability analysis |
| Markov | atacs | Use atacs steady-state Markov analysis engine |
| Markov | ctmc-transient | Transient distribution analysis |
| Field | Description |
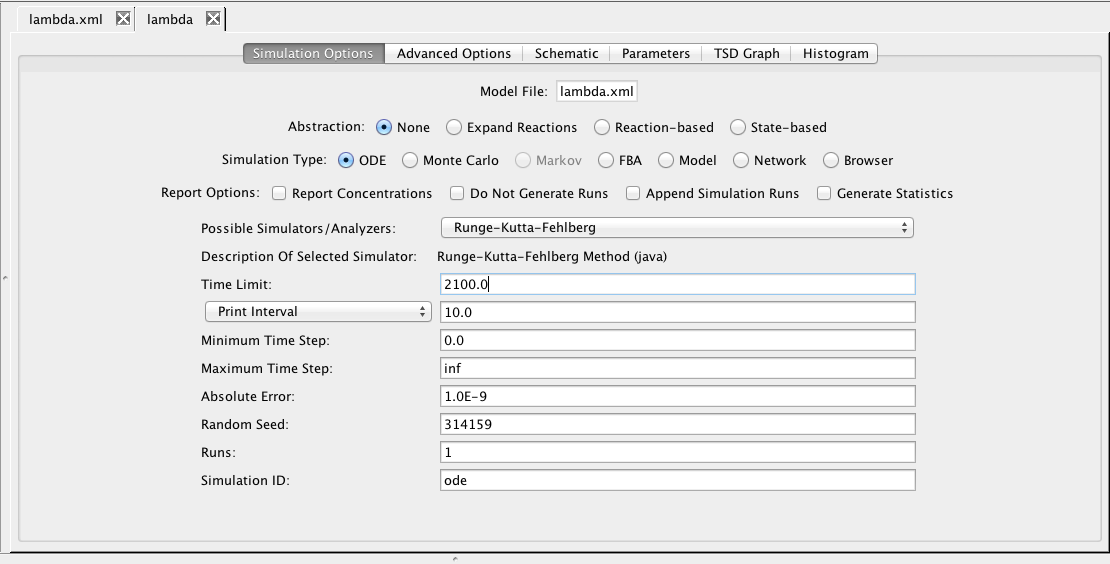
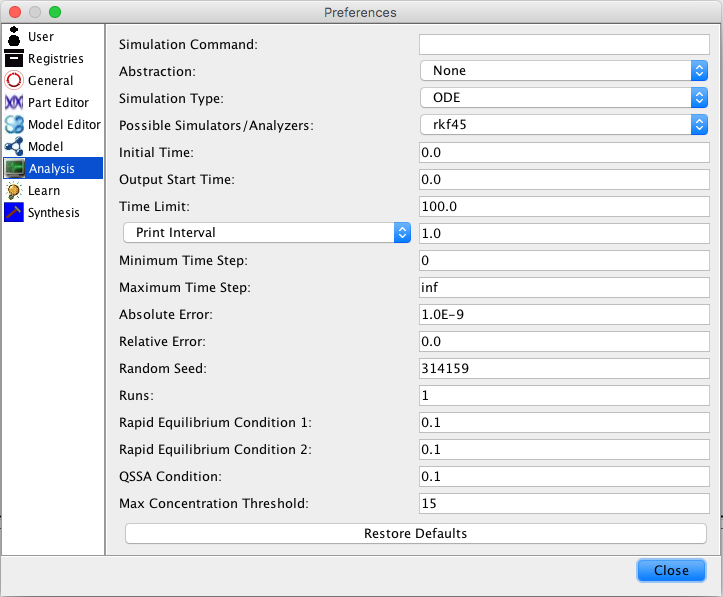
| Time Limit | The simulation time limit |
| Choose one: | |
| Print Interval | The print time interval for each simulation run |
| Minimum Print Interval | Print on change but no more often than this interval |
| Number of Steps | Number of steps to print |
| Minimum Time Step | The smallest time step allowed |
| Maximum Time Step | The largest time step allowed |
| (also the time step used for the Euler method) | |
| Absolute Error | Used by the adaptive time step ODE methods |
| Random Seed | An integer number as a seed to generate random numbers |
| Runs | The number of random simulation runs to perform |
| Simulation ID | Creates a simulation directory with the ID name |
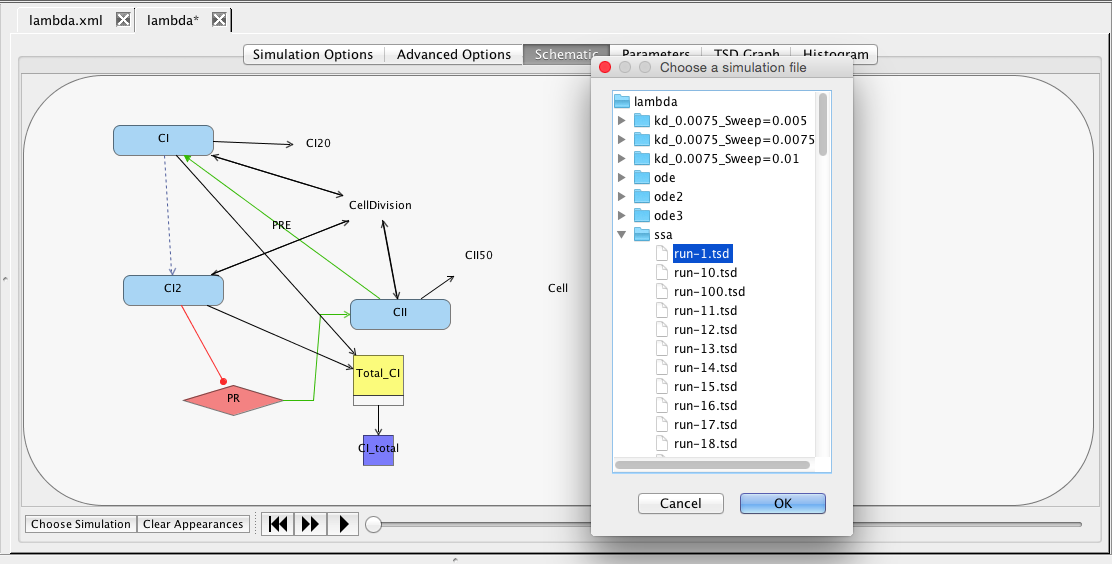
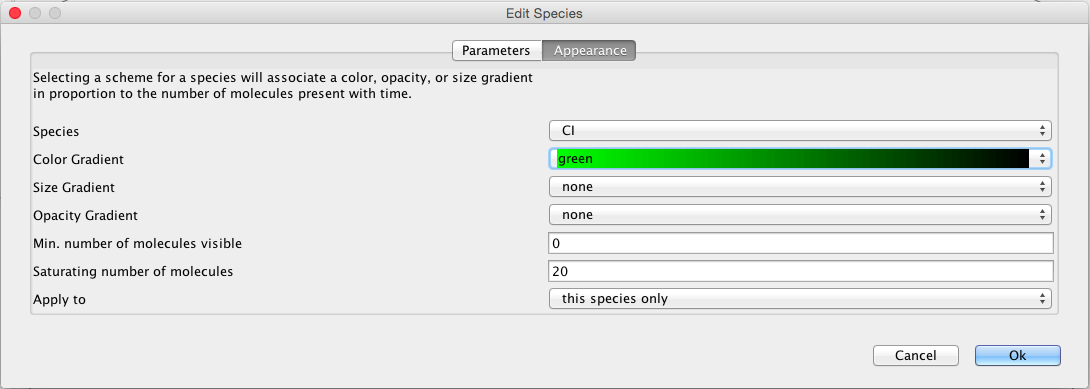
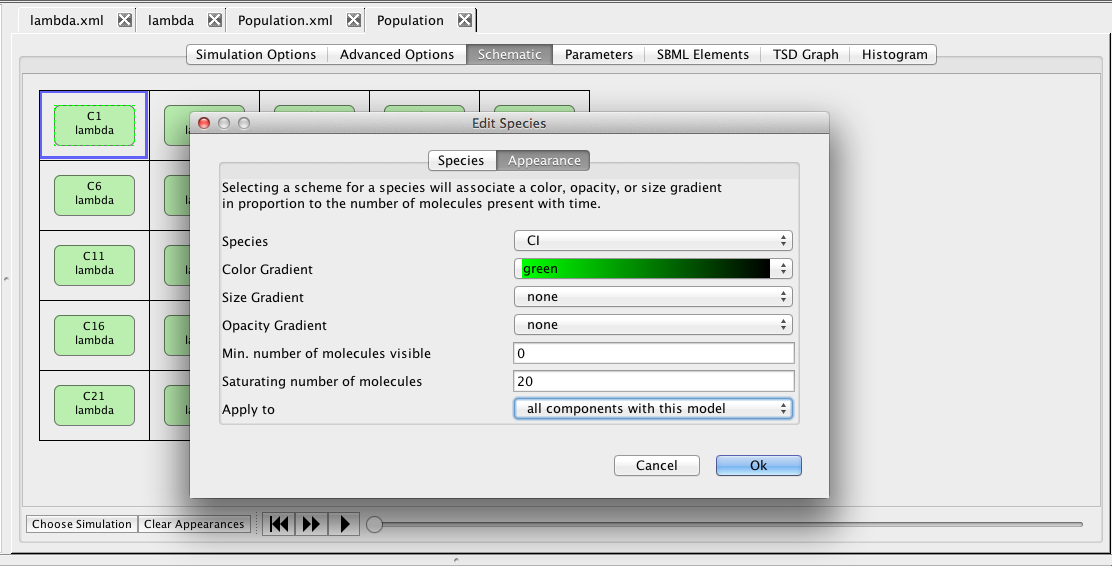
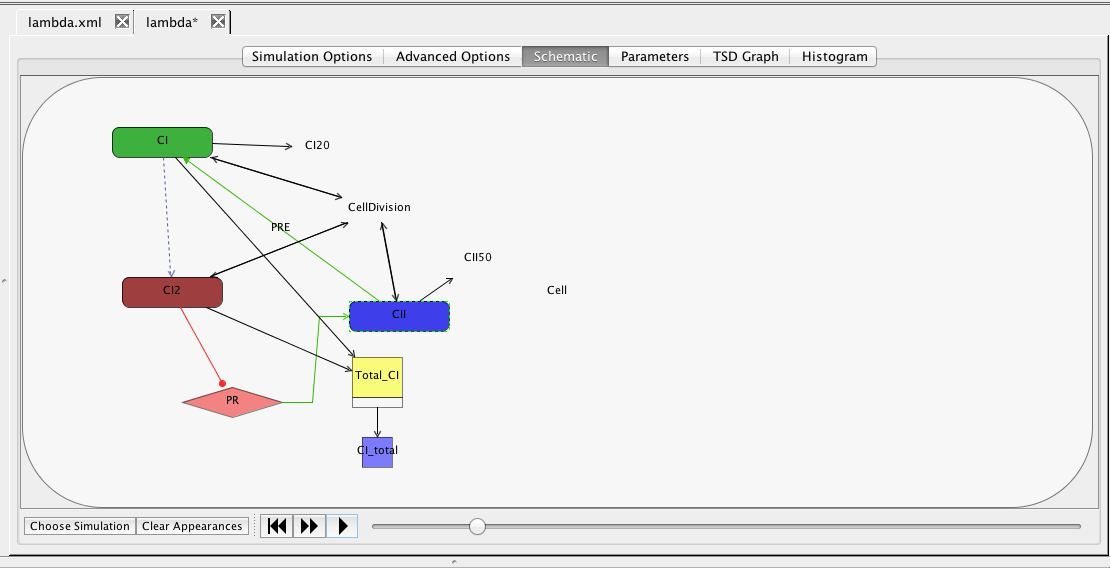
 | 
|
 | 
|
 | 
|